
Candida glabrata: Pathogenicity and Resistance Mechanisms for Adaptation and Survival


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Candida glabrata Virulence Factors
2.1. Enzyme Secretion
2.2. Adhesin Cell-Like Protein
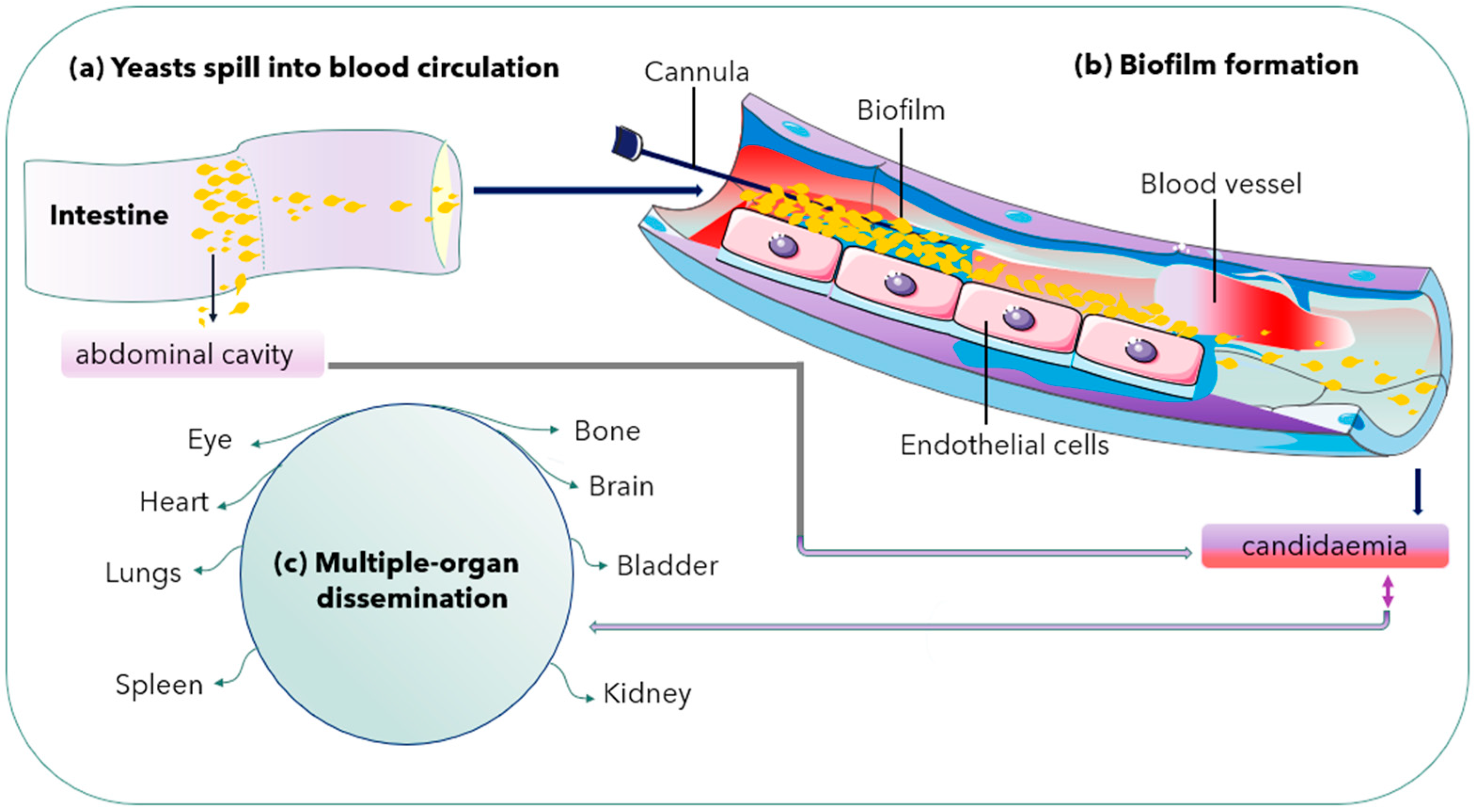
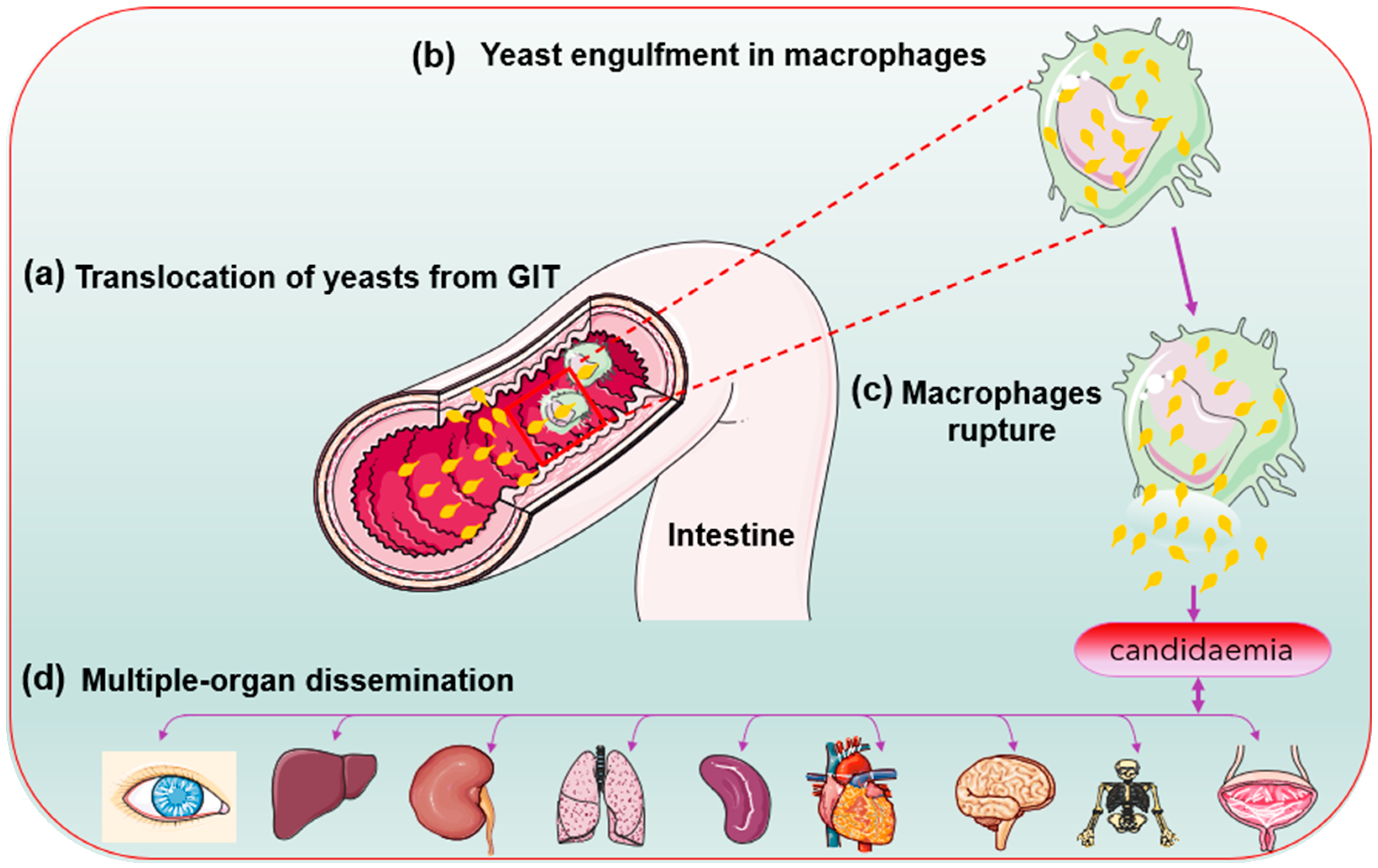
2.3. Biofilm Formation
2.4. Presence of a Stable Cell Wall
2.5. Novel Hybrid Iron Regulation and Acquisition Strategies
2.6. Adaptation to Various Environmental Conditions
2.7. Replicative Ageing
3. Drug-Resistance Mechanisms of Candida glabrata
3.1. Types of Drug Resistance Mechanisms
3.1.1. Azole Resistance
3.1.2. Echinocandins Resistance
3.1.3. Polyenes Resistance
4. Conclusions and Way Forward
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pappas, P.G.; Lionakis, M.S.; Arendrup, M.C.; Ostrosky-Zeichner, L.; Kullberg, B.J. Invasive candidiasis. Nat. Rev. Dis. Primers 2018, 4, 18026. [Google Scholar] [CrossRef] [PubMed]
- Won, E.J.; Shin, J.H.; Choi, M.J.; Lee, W.G.; Park, Y.-J.; Uh, Y.; Kim, S.-Y.; Lee, M.-K.; Kim, S.H.; Shin, M.G.; et al. Antifungal Susceptibilities of Bloodstream Isolates of Candida Species from Nine Hospitals in Korea: Application of New Antifungal Breakpoints and Relationship to Antifungal Usage. PLoS ONE 2015, 10, e0118770. [Google Scholar] [CrossRef]
- Timmermans, B.; De Las Peñas, A.; Castaño, I.; Van Dijck, P. Adhesins in Candida glabrata. J. Fungi 2018, 4, 60. [Google Scholar] [CrossRef]
- Norimatsu, Y.; Morii, D.; Kogure, A.; Hamanaka, T.; Kuwano, Y.; Yokozawa, T.; Oda, T. A case of breakthrough Candida parapsilosis fungemia during micafungin therapy for a Candida glabrata bloodstream infection. Med. Mycol. Case Rep. 2017, 16, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Vanhee, L.M.E.; Meersseman, W.; Lagrou, K.; Maertens, J.; Nelis, H.J.; Coenye, T. Rapid and Direct Quantification of Viable Candida Species in Whole Blood by Use of Immunomagnetic Separation and Solid-Phase Cytometry. J. Clin. Microbiol. 2010, 48, 1126–1131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lamagni, M.T.L.; Evans, B.G.; Shigematsu, M.; Johnson, E.M. Emerging trends in the epidemiology of invasive mycoses in England and Wales (1990–1999). Epidemiol. Infect. 2001, 126, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, Candida parapsilosis and Candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Kounatidis, I.; Ames, L.; Mistry, R.; Ho, H.-L.; Haynes, K.; Ligoxygakis, P. A Host-Pathogen Interaction Screen Identifies ada2 as a Mediator of Candida glabrata Defenses against Reactive Oxygen Species. G3 Genes Genomes Genet. 2018, 8, 1637–1647. [Google Scholar] [CrossRef]
- Kumar, K.; Askari, F.; Sahu, M.S.; Kaur, R. Candida glabrata: A Lot More Than Meets the Eye. Microorganisms 2019, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Husain, A.; Khan, S.; Mujeeb, M.; Bhandari, A. Design, synthesis, molecular properties and antimicrobial activities of some novel 2(3H) pyrrolone derivatives. J. Saudi Chem. Soc. 2015, 19, 340–346. [Google Scholar] [CrossRef]
- McCarty, T.P.; Pappas, P.G. Invasive Candidiasis. Infect. Dis. Clin. N. Am. 2016, 30, 103–124. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D. Epidemiology of Invasive Candidiasis: A Persistent Public Health Problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Galocha, M.; Pais, P.; Cavalheiro, M.; Pereira, D.; Viana, R.; Teixeira, M.C. Divergent approaches to virulence in C. Albicans and C. Glabrata: Two sides of the same coin. Int. J. Mol. Sci. 2019, 20, 2345. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torrado, R.; Querol, A. Saccharomyces cerevisiae show low levels of traversal across the human blood brain barrier in vitro. F1000Research 2017, 6, 944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, S.; Pan, A.; Li, J.; Liu, B. Surveillance of antifungal susceptibilities in clinical isolates of Candida species at 36 hospitals in China from 2009 to 2013. Int. J. Infect. Dis. 2015, 33, 1–4. [Google Scholar] [CrossRef]
- Kaur, R.; Domergue, R.; Zupancic, M.L.; Cormack, B.P. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 2005, 8, 378–384. [Google Scholar] [CrossRef]
- Singh, R.; Parija, S. Candida parapsilosis: An emerging fungal pathogen. Indian J. Med. Res. 2012, 136, 671–673. [Google Scholar]
- Gabaldón, T.; Carreté, L. The birth of a deadly yeast: Tracing the evolutionary emergence of virulence traits in Candida gla-brata. FEMS Yeast Res. 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Moreira, A.; Silva, S.; Botelho, C.; Sampaio, P.; Pais, C.; Henriques, M. Candida bracarensis: Evaluation of Virulence Factors and its Tolerance to Amphotericin B and Fluconazole. Mycopathologia 2015, 180, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.; Netea, M.G.; Kullberg, B.J. Patient Susceptibility to Candidiasis—A Potential for Adjunctive Immunotherapy. J. Fungi 2018, 4, 9. [Google Scholar] [CrossRef]
- Kasper, L.; Seider, K.; Gerwien, F.; Allert, S.; Brunke, S.; Schwarzmüller, T.; Ames, L.; Zubiria-Barrera, C.; Mansour, M.K.; Becken, U.; et al. Identification of Candida glabrata Genes Involved in pH Modulation and Modification of the Phagosomal Environment in Macrophages. PLoS ONE 2014, 9, e96015. [Google Scholar] [CrossRef]
- Schwarzmüller, T.; Ma, B.; Hiller, E.; Istel, F.; Tscherner, M.; Brunke, S.; Ames, L.; Firon, A.; Green, B.; Cabral, V.; et al. Systematic Phenotyping of a Large-Scale Candida glabrata Deletion Collection Reveals Novel Antifungal Tolerance Genes. PLoS Pathog. 2014, 10, e1004211. [Google Scholar] [CrossRef]
- Linde, J.; Duggan, S.; Weber, M.; Horn, F.; Sieber, P.; Hellwig, D.; Riege, K.; Marz, M.; Martin, R.; Guthke, R.; et al. Defining the transcriptomic landscape of Candida glabrata by RNA-Seq. Nucleic Acids Res. 2015, 43, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Rapala-Kozik, M.; Bochenska, O.; Zajac, D.; Karkowska-Kuleta, J.; Gogol, M.; Zawrotniak, M.; Kozik, A. Extracellular proteinases of Candida species pathogenic yeasts. Mol. Oral Microbiol. 2018, 33, 113–124. [Google Scholar] [CrossRef]
- Shang-Jie, Y.; Ya-Lin Chang, Y.-L.C. Deletion of ADA2 Increases Antifungal Drug Susceptibility and Virulence in Candida glabrata. Antimicrob. Agents Chemother. 2018, 62, e01924-17. [Google Scholar]
- Kawai, A.; Yamagishi, Y.; Mikamo, H. In vitro efficacy of liposomal amphotericin B, micafungin and fluconazole against non-albicans Candida species biofilms. J. Infect. Chemother. 2015, 21, 647–653. [Google Scholar] [CrossRef]
- Brunke, S.; Hube, B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, M.S.; Binkley, J.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database (CGD): Incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017, 45, D592–D596. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Latha, R.; Vedhagiri, K.; Sathiamoorthi, T.; Jayarani, G.; Sasikala, R.; Selvin, J.; Natarajaseenivasan, K. Phospholipase C, proteinase and hemolytic activities of Candida spp. isolated from pulmonary tuberculosis patients. J. Mycol. Med. 2009, 19, 3–10. [Google Scholar] [CrossRef]
- Nahas, J.V.; Iosue, C.L.; Shaik, N.F.; Selhorst, K.; He, B.Z.; Wykoff, D.D. Dynamic changes in yeast phosphatase families allow for specialization in phosphate and thiamine starvation. G3 Genes Genomes Genet. 2018, 8, 2333–2343. [Google Scholar] [CrossRef]
- Figueiredo-Carvalho, M.H.; Ramos, L.; Barbedo, L.; Chaves, A.L.D.S.; Muramoto, I.A.; Dos Santos, A.L.S.; Almeida-Paes, R.; Zancopé-Oliveira, R.M. First description of Candida nivariensis in Brazil: Antifungal susceptibility profile and potential virulence attributes. Mem. Inst. Oswaldo Cruz 2016, 111, 51–58. [Google Scholar] [CrossRef]
- Kühbacher, A.; Burger-Kentischer, A.; Rupp, S. Interaction of Candida Species with the Skin. Microorganisms 2017, 5, 32. [Google Scholar] [CrossRef]
- Hoyer, L.L.; Green, C.B.; Oh, S.; Zhao, X. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family a sticky pursuit. Med. Mycol. 2008, 46, 1–15. [Google Scholar] [CrossRef]
- De Groot, P.W.J.; Kraneveld, E.A.; Qing, Y.Y.; Dekker, H.L.; Groß, U.; Crielaard, W.; de Koster, C.G.; Bader, O.; Klis, F.M.; Weig, M. The cell wall of the human pathogen Candida glabrata: Differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell 2008, 7, 1951–1964. [Google Scholar] [CrossRef]
- Kohlenberg, A.; Struelens, M.J.; Monnet, D.L.; Plachouras, D.; The Candida Auris Survey Collaborative Group. Candida auris: Epidemiological situation, laboratory capacity and preparedness in European Union and European economic area countries, 2013 to 2017. Eurosurveillance 2018, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, M.; Posteraro, B.; Trecarichi, E.M.; Fiori, B.; Rossi, M.; Porta, R.; de Gaetano Donati, K.; La Sorda, M.; Spanu, T.; Fadda, G.; et al. Biofilm production by Candida species and in-adequate antifungal therapy as predictors of mortality for patients with candidemia. J. Clin. Microbiol. 2007, 45, 1843–1850. [Google Scholar] [CrossRef]
- Nett, J.E.; Andes, D.R. Contributions of the Biofilm Matrix to Candida Pathogenesis. J. Fungi 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.G.; Uppuluri, P.; Tristan, A.R.; Wormley, F.L.; Mowat, E.; Ramage, G.; Lopez-Ribot, J.L. A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat. Protoc. 2008, 3, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.S.; Lana, D.F.D.; Mezzari, A. Candida auris: Emergence and Epidemiology of a Highly Pathogenic Yeast. Clin. Biomed. Res. 2017, 37, 247–254. [Google Scholar] [CrossRef][Green Version]
- Mota, S.; Alves, R.; Carneiro, C.; Silva, S.; Brown, A.J.; Istel, F.; Kuchler, K.; Sampaio, P.; Casal, M.; Henriques, M.; et al. Candida glabrata susceptibility to antifungals and phagocytosis is modulated by acetate. Front. Microbiol. 2015, 6, 919. [Google Scholar] [CrossRef] [PubMed]
- Sardi, J.C.O.; Scorzoni, L.; Bernardi, T.; Fusco-Almeida, A.M.; Giannini, M.J.M. Candida species: Current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 2013, 62, 10–24. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Patterson, T.F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J. Infect. Dis. 2017, 216, S445–S451. [Google Scholar] [CrossRef]
- Dantas, A.D.S.; Lee, K.K.; Raziunaite, I.; Schaefer, K.; Wagener, J.; Yadav, B.; Gow, N.A. Cell biology of Candida albicans-host interactions. Curr. Opin. Microbiol. 2016, 34, 111–118. [Google Scholar] [CrossRef]
- Jeffery-Smith, A.; Taori, S.K.; Schelenz, S.; Jeffery, K.; Johnson, E.M.; Borman, A.; Manuel, R.; Brown, C.S. Candida auris: A Review of the Literature. Clin. Microbiol. Rev. 2018, 31, e00029-17. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhaheri, R.S.; Douglas, L.J. Absence of Amphotericin B-Tolerant Persister Cells in Biofilms of Some Candida Species. Antimicrob. Agents Chemother. 2008, 52, 1884–1887. [Google Scholar] [CrossRef]
- Charlet, R.; Pruvost, Y.; Tumba, G.; Istel, F.; Poulain, D.; Kuchler, K.; Sendid, B.; Jawhara, S. Remodeling of the Candida glabrata cell wall in the gastrointestinal tract affects the gut microbiota and the immune response. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Van Der Meer, J.W.M.; Kullberg, B.-J.; Van De Veerdonk, F.L. Immune defence against Candida fungal infections. Nat. Rev. Immunol. 2015, 15, 630–642. [Google Scholar] [CrossRef]
- Molina, M.; Gil, C.; Pla, J.; Arroyo, J.; Nombela, C. Protein Localisation Approaches for Understanding Yeast Cell Wall Biogenesis. Histol. Stud. Yeast 2000, 612, 601–612. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Suneetha, K.J.; Kaur, R. A systematic analysis reveals an essential role for high-affinity iron uptake system, haemolysin and CFEM domain-containing protein in iron homoeostasis and virulence in Candida glabrata. Biochem. J. 2014, 463, 103–114. [Google Scholar] [CrossRef]
- Perlin, D.S. Echinocandin resistance, susceptibility testing and prophylaxis: Implications for patient management. Drugs 2014, 74, 1573–1585. [Google Scholar] [CrossRef]
- Enkler, L.; Richer, D.; Marchand, A.L.; Ferrandon, D.; Jossinet, F. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci. Rep. 2016, 6, 35766. [Google Scholar] [CrossRef] [PubMed]
- Gerwien, F.; Safyan, A.; Wisgott, S.; Hille, F.; Kaemmer, P.; Linde, J.; Brunke, S.; Kasper, L.; Hube, B. A Novel Hybrid Iron Regulation Network Combines Features from Pathogenic and Nonpathogenic Yeasts. mBio 2016, 7, e01782-16. [Google Scholar] [CrossRef]
- Haas, H.; Eisendle, M.; Turgeon, B.G. Siderophores in Fungal Physiology and Virulence. Annu. Rev. Phytopathol. 2008, 46, 149–187. [Google Scholar] [CrossRef] [PubMed]
- Ehrensberger, K.M.; Bird, A.J. Hammering out details: Regulating metal levels in eukaryotes. Trends Biochem. Sci. 2011, 36, 524–531. [Google Scholar] [CrossRef]
- De Wever, V.; Reiter, W.; Ballarini, A.; Ammerer, G.; Brocard, C. A dual role for PP1 in shaping the Msn2-dependent transcrip-tional response to glucose starvation. EMBO J. 2005, 24, 4115–4123. [Google Scholar] [CrossRef]
- Roetzer, A.; Gregori, C.; Jennings, A.M.; Quintin, J.; Ferrandon, D.; Butler, G.; Kuchler, K.; Ammerer, G.; Schüller, C. Candida glabrata environmental stress response involves Saccharomyces cerevisiae Msn2/4 orthologous transcription factors. Mol. Microbiol. 2008, 69, 603–620. [Google Scholar] [CrossRef]
- Wu, J.; Chen, X.; Cai, L.; Tang, L.; Liu, L. Transcription factors Asg1p and Hal9p regulate pH homeostasis in Candida glabrata. Front. Microbiol. 2015, 6, 843. [Google Scholar] [CrossRef]
- Dadar, M.; Tiwari, R.; Karthik, K.; Chakraborty, S.; Shahali, Y.; Dhama, K. Candida albicans—Biology, molecular characterization, pathogenicity, and advances in diagnosis and control—An update. Microb. Pathog. 2018, 117, 128–138. [Google Scholar] [CrossRef]
- Chew, S.Y.; Chee, W.J.Y.; Than, L.T.L. The glyoxylate cycle and alternative carbon metabolism as metabolic adaptation strategies of Candida glabrata: Perspectives from Candida albicans and Saccharomyces cerevisiae. J. Biomed. Sci. 2019, 26, 1–10. [Google Scholar] [CrossRef]
- Ullah, A.; Lopes, M.I.; Brul, S.; Smits, G. Intracellular pH homeostasis in Candida glabrata in infection-associated conditions. Microbiology 2013, 159, 803–813. [Google Scholar] [CrossRef]
- Ho, H.-L.; Haynes, K.F. Candida glabrata: New tools and technologies—Expanding the toolkit. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef]
- Chew, S.Y.; Ho, K.L.; Cheah, Y.K.; Sandai, D.; Brown, A.J.; Than, L.T.L. Physiologically Relevant Alternative Carbon Sources Modulate Biofilm Formation, Cell Wall Architecture, and the Stress and Antifungal Resistance of Candida glabrata. Int. J. Mol. Sci. 2019, 20, 3172. [Google Scholar] [CrossRef]
- Huang, M.; Khan, J.; Kaur, M.; Vanega, J.D.T.; Patiño, O.A.A.; Ramasubramanian, A.K.; Kao, K.C. CgSTE11 mediates cross tolerance to multiple environmental stressors in Candida glabrata. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Roetzer, A.; Gabaldón, T.; Schüller, C. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: Important adaptations for an opportunistic pathogen. FEMS Microbiol. Lett. 2010, 314, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Lin, X.; Qi, Y.; Liu, H.; Chen, X.; Liu, L.; Chen, J. Crz1p Regulates pH Homeostasis in Candida glabrata by Altering Membrane Lipid Composition. Appl. Environ. Microbiol. 2016, 82, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.; Pedro, N.A.A.; Salazar, S.B.; Mira, N.P. Effect of Acetic Acid and Lactic Acid at Low pH in Growth and Azole Resistance of Candida albicans and Candida glabrata. Front. Microbiol. 2019, 9, 3265. [Google Scholar] [CrossRef]
- Yih, C.S.; Than, L.; Lung, T. Candida glabrata: Niche adaptation and mechanisms of survival. In Updates in Medical Microbiology; Universiti Putra Malaysia Press: Selangor, Malaysia, 2012. [Google Scholar]
- Roetzer, A.; Gratz, N.; Kovarik, P.; Schüller, C. Autophagy supports Candida glabrata survival during phagocytosis. Cell. Microbiol. 2010, 12, 199–216. [Google Scholar] [CrossRef]
- Del Poeta, M. Role of phagocytosis in the virulence of Cryptococcus neoformans. Eukaryot. Cell. 2004, 3, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.Y.; Ho, K.L.; Cheah, Y.K.; Ng, T.S.; Sandai, D.; Brown, A.J.P.; Than, L.T.L. Glyoxylate cycle gene ICL1 is essential for the metabolic flexibility and virulence of Candida glabrata. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Jayampath Seneviratne, C.; Wang, Y.; Jin, L.; Abiko, Y.; Samaranayake, L.P. Proteomics of drug resistance in Candida glabrata biofilms. Proteomics 2010, 10, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves Dos Santos, M.T.P.; Benito, M.J.; Córdoba, M.D.G.; Alvarenga, N.; de Herrera, S.R.-M.S. Yeast community in traditional Portuguese Serpa cheese by culture-dependent and -independent DNA approaches. Int. J. Food Microbiol. 2017, 262, 63–70. [Google Scholar] [CrossRef]
- Bouklas, T.; Alonso-Crisóstomo, L.; Székely, T.; Navarro, E.D.; Orner, E.P.; Smith, K.; Munshi, M.A.; Del Poeta, M.; Balazsi, G.; Fries, B.C. Generational distribution of a Candida glabrata population: Resilient old cells prevail, while younger cells dominate in the vulnerable host. PLoS Pathog. 2017, 13, e1006355. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Bouklas, T.; Fries, B.C. Replicative Aging in Pathogenic Fungi. J. Fungi 2020, 7, 6. [Google Scholar] [CrossRef]
- Bhattacharya, S.A.; Fries, B.C. Enhanced Efflux Pump Activity in Old Candida glabrata Cells. Antimicrob. Agents Chemother. 2018, 62, e02227-17. [Google Scholar] [CrossRef]
- Alastruey-Izquierdo, A.; Melhem, M.S.C.; Bonfietti, L.X.; Rodriguez-Tudela, J.L. Susceptibility Test for Fungi: Clinical and Laboratorial Correlations in Medical Mycology. Rev. Inst. Med. Trop. Sao Paulo 2015, 57, 57–64. [Google Scholar] [CrossRef]
- Costa, C.; Ribeiro, J.; Miranda, I.M.; Silva-Dias, A.; Cavalheiro, M.; Costa-de-Oliveira, S.; Rodrigues, A.G.; Teixeira, M.C. Clotrimazole drug resistance in Candida glabrata clinical isolates correlates with increased expression of the drug: H+ antiporters CgAqr1, CgTpo1_1, CgTpo3, and CgQdr2. Front. Microbiol. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Irinyi, L.; Lackner, M.; de Hoog, G.S.; Meyer, W. DNA barcoding of fungi causing infections in humans and animals. Fungal Biol. 2016, 120, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Whaley, S.G.; Berkow, E.L.; Rybak, J.M.; Nishimoto, A.T.; Barker, K.S.; Rogers, P.D. Azole Antifungal Resistance in Candida albicans and Emerging Non-albicans Candida Species. Front. Microbiol. 2017, 7, 2173. [Google Scholar] [CrossRef]
- Yoo, J.I.; Choi, C.W.; Lee, K.M.; Lee, Y.S. Gene Expression and Identification Related to Fluconazole Resistance of Candida glabrata Strains. Osong Public Health Res. Perspect. 2010, 1, 36–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jensen, R.H.; Johansen, H.K.; Søes, L.M.; Lemming, L.E.; Rosenvinge, F.S.; Nielsen, L.; Olesen, B.; Kristensen, L.; Dzajic, E.; Astvad, K.; et al. Posttreatment Antifungal Resistance among Colonizing Candida Isolates in Candidemia Patients: Results from a Systematic Multicenter Study. Antimicrob. Agents Chemother. 2016, 60, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Chen, S.-A.; Halliday, C.; Kennedy, K.; Playford, E.; Marriott, D.; Slavin, M.; Sorrell, T.; Sintchenko, V. Identification of genetic markers of resistance to echinocandins, azoles and 5-fluorocytosine in Candida glabrata by next-generation sequencing: A feasibility study. Clin. Microbiol. Infect. 2017, 23, 676.e7–676.e10. [Google Scholar] [CrossRef] [PubMed]
- Basas, J.; Palau, M.; Gomis, X.; Almirante, B.; Gavaldà, J. Efficacy of liposomal amphotericin B and anidulafungin using an an-tifungal lock technique (ALT) for catheter-related Candida albicans and Candida glabrata infections in an experimental model. PLoS ONE 2019, 14, e0212426. [Google Scholar] [CrossRef]
- Tsui, C.K.M.; Woodhall, J.; Chen, W.; André Lévesque, C.; Lau, A.; Schoen, C.D.; Baschien, C.; Najafzadeh, M.J.; de Hoog, G.S. Molecular techniques for pathogen identification and fungus detection in the environment. IMA Fungus 2011, 2, 177–189. [Google Scholar] [CrossRef]
- Gohar, A.A.; Badali, H.; Shokohi, T.; Nabili, M.; Amirrajab, N.; Moazeni, M. Expression patterns of ABC transporter genes in fluconazole-resistant Candida glabrata. Mycopathologia 2017, 182, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and management. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Farahyar, S.; Zaini, F.; Kordbacheh, P.; Rezaie, S.; Falahati, M.; Safara, M.; Raoofian, R.; Hatami, K.; Mohebbi, M.; Heidari, M. Expression of Efflux Pumps and Fatty Acid Activator One Genes in Azole Resistant Candida glabrata Isolated from Immunocompromised Patients. Acta Med. Iran. 2016, 54, 458–464. [Google Scholar] [PubMed]
- Berkow, E.L.; Lockhart, S.R. Activity of CD101, a long-acting echinocandin, against clinical isolates of Candida auris. Diagn. Microbiol. Infect. Dis. 2018, 90, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Poláková, S.; Blume, C.; Zárate, J.Á.; Mentel, M.; Jørck-Ramberg, D.; Stenderup, J.; Piškur, J. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc. Natl. Acad. Sci. USA 2009, 106, 2688–2693. [Google Scholar] [CrossRef]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.; Boselli, M.; Dunham, M.; Amon, A. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Krol, A.A.; Sarti, K.E.; Bennett, J.E. Candida glabrata PDR1, a Transcriptional Regulator of a Pleiotropic Drug Resistance Network, Mediates Azole Resistance in Clinical Isolates and Petite Mutants. Antimicrob. Agents Chemother. 2006, 50, 1384–1392. [Google Scholar] [CrossRef]
- Ferrari, S.; Sanguinetti, M.; De Bernardis, F.; Torelli, R.; Posteraro, B.; Vandeputte, P.; Sangland, D. Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrob. Agents Chemother. 2011, 55, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Nedret Koç, A.; Gökahmetòǧlu, S.; Òǧuzkaya, M. Comparison of Etest with the broth microdilution method in susceptibility testing of yeast isolates against four antifungals. Mycoses 2000, 43, 293–297. [Google Scholar] [CrossRef]
- Song, Y.B.; Suh, M.K.; Ha, G.Y.; Kim, H. Antifungal Susceptibility Testing with Etest for Candida Species Isolated from Patients with Oral Candidiasis. Ann. Dermatol. 2015, 27, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Sri, J.B.; Premamalini, T.; Rajyoganandh, S.V.; Anupma, J.K. Pattern of susceptibility to azoles by E test method in candidemia patients. Int. J. Res. Med. Sci. 2015, 3, 2118–2122. [Google Scholar]
- Chen, M.; Xu, Y.; Hong, N.; Yang, Y.; Lei, W.; Du, L.; Zhao, J.; Lei, X.; Xiong, L.; Cai, L.; et al. Epidemiology of fungal infections in China. Front. Med. 2018, 12, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Larkin, E.L.; Dharmaiah, S.; Ghannoum, M.A. Biofilms and beyond: Expanding echinocandin utility. J. Antimicrob. Chemother. 2018, 73, 73–81. [Google Scholar] [CrossRef]
- Wayne, P. Clinical and laboratory standards institute: Reference method for broth dilution antifungal susceptibility testing of yeasts. In Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; p. 30. [Google Scholar]
- Wanjare, S.; Gupta, R.; Mehta, P. Caspofungin MIC Distribution amongst Commonly Isolated Candida Species in a Tertiary Care Centre—An Indian Experience. J. Clin. Diagn. Res. 2016, 10, DC11–DC13. [Google Scholar] [CrossRef]
- Colombo, A.L.; Júnior, J.N.D.A.; Guinea, J. Emerging multidrug-resistant Candida species. Curr. Opin. Infect. Dis. 2017, 30, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, S.F.; Lee, T.J.; Chew, K.L.; Lin, S.; Yeo, D.; Setia, S. Echinocandins for management of invasive candidiasis in patients with liver disease and liver transplantation. Infect. Drug Resist. 2018, 11, 805–819. [Google Scholar] [CrossRef]
- Sasso, M.; Roger, C.; Lachaud, L. Rapid emergence of FKS mutations in Candida glabrata isolates in a peritoneal candidiasis. Med. Mycol. Case Rep. 2017, 16, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M. Methods in Molecular Biology. TM Series Editor. Life Sci. 2009, 531, 588. [Google Scholar]
- Shields, R.K.; Nguyen, M.H.; Press, E.G.; Cumbie, R.; Driscoll, E.; Pasculle, A.W.; Clancy, C.J. Rate of FKS Mutations among Consecutive Candida Isolates Causing Bloodstream Infection. Antimicrob. Agents Chemother. 2016, 60, 1954. [Google Scholar] [CrossRef]
- Aslani, N.; Janbabaei, G.; Abastabar, M.; Meis, J.F.; Babaeian, M.; Khodavaisy, S.; Boekhout, T.; Badali, H. Identification of uncommon oral yeasts from cancer patients by MALDI-TOF mass spectrometry. BMC Infect. Dis. 2018, 18, 24. [Google Scholar] [CrossRef]
- Rodrigues, C.F.; Silva, S.; Azeredo, J.; Henriques, M. Candida glabrata’s recurrent infections: Biofilm formation during Amphotericin B treatment. Lett. Appl. Microbiol. 2016, 63, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Grela, E.; Piet, M.; Luchowski, R.; Grudzinski, W.; Paduch, R.; Gruszecki, W.I. Imaging of human cells exposed to an antifungal antibiotic amphotericin B reveals the mechanisms associated with the drug toxicity and cell defence. Sci. Rep. 2018, 8, 14067. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris article. Emerg. Microb. Infect. 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Tay, S.T.; Lotfalikhani, A.; Sabet, N.S.; Ponnampalavanar, S.; Sulaiman, S.; Na, S.L.; Ng, K.P. Occurrence and Characterization of Candida nivariensis from a Culture Collection of Candida glabrata Clinical Isolates in Malaysia. Mycopathologia 2014, 178, 307–314. [Google Scholar] [CrossRef]
- Shokohi, T.; Aslani, N.; Ahangarkani, F.; Meyabadi, M.F.; Hagen, F.; Meis, J.F.; Boekhout, T.; Kolecka, A.; Badali, H. Candida infanticola and Candida spencermartinsiae yeasts: Possible emerging species in cancer patients. Microb. Pathog. 2018, 115, 353–357. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, Y.; Chew, S.Y.; Than, L.T.L. Candida glabrata: Pathogenicity and Resistance Mechanisms for Adaptation and Survival. J. Fungi 2021, 7, 667. https://doi.org/10.3390/jof7080667
Hassan Y, Chew SY, Than LTL. Candida glabrata: Pathogenicity and Resistance Mechanisms for Adaptation and Survival. Journal of Fungi. 2021; 7(8):667. https://doi.org/10.3390/jof7080667
Chicago/Turabian StyleHassan, Yahaya, Shu Yih Chew, and Leslie Thian Lung Than. 2021. "Candida glabrata: Pathogenicity and Resistance Mechanisms for Adaptation and Survival" Journal of Fungi 7, no. 8: 667. https://doi.org/10.3390/jof7080667
APA StyleHassan, Y., Chew, S. Y., & Than, L. T. L. (2021). Candida glabrata: Pathogenicity and Resistance Mechanisms for Adaptation and Survival. Journal of Fungi, 7(8), 667. https://doi.org/10.3390/jof7080667