
Histoplasma capsulatum Isolated from Tadarida brasiliensis Bats Captured in Mexico Form a Sister Group to North American Class 2 Clade

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
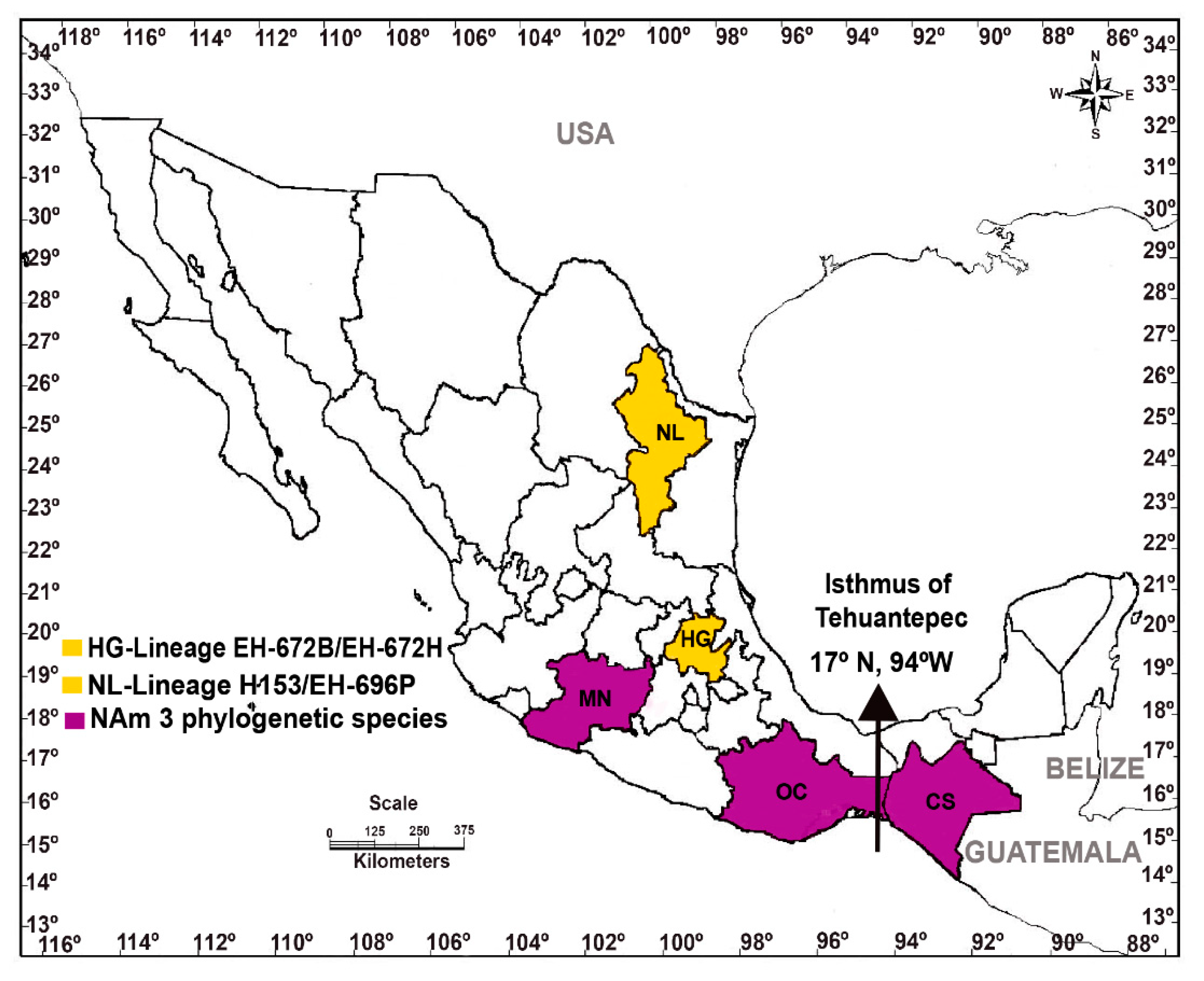
2.1. New Histoplasma capsulatum Isolates Studied
2.2. Histoplasma capsulatum Sequences
2.3. DNA Extraction, PCR, and Sequencing of Histoplasma capsulatum Isolates
2.4. Histoplasma capsulatum Sequence Alignments and BLASTn Analyses
2.5. Congruence Analysis
2.6. Phylogenetic Reconstruction
2.7. Coalescence Analysis
2.8. Concatenated Sequence-Types (CSTs) Network
2.9. Nucleotide Diversity (π)
3. Results
3.1. Histoplasma capsulatum BLASTn Analysis
3.2. Congruence Analysis
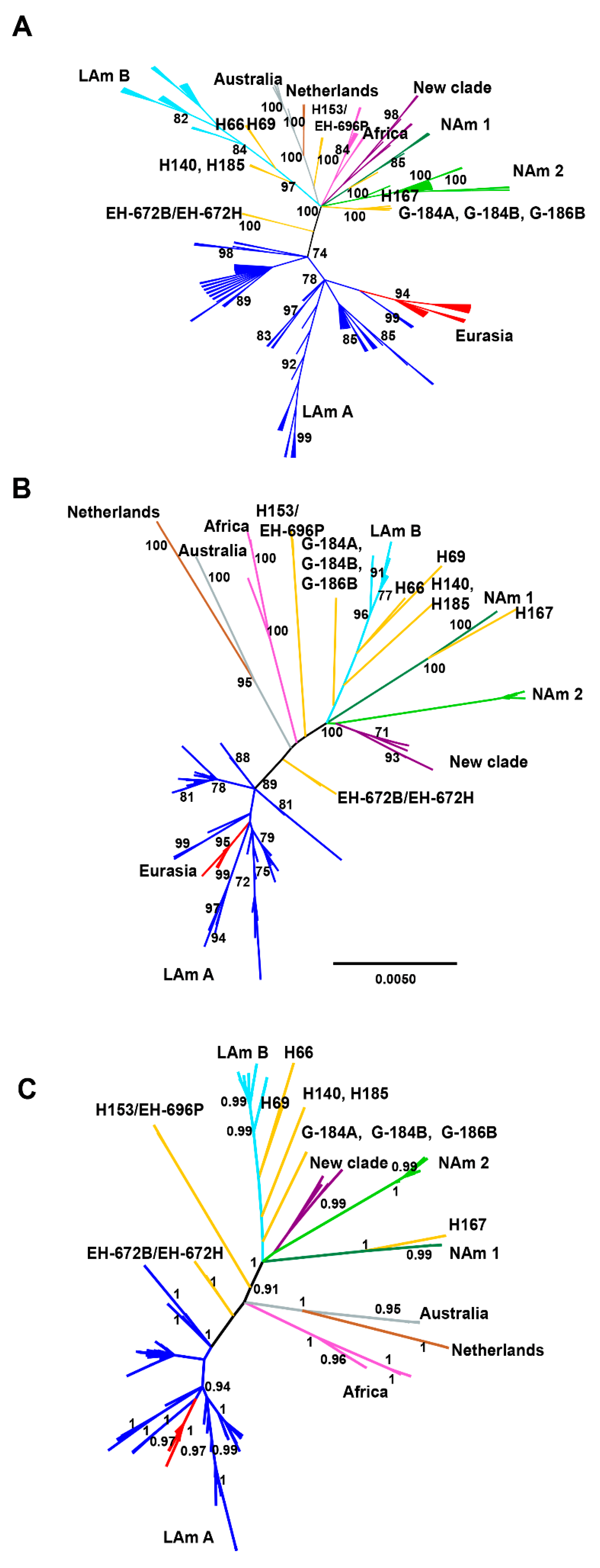
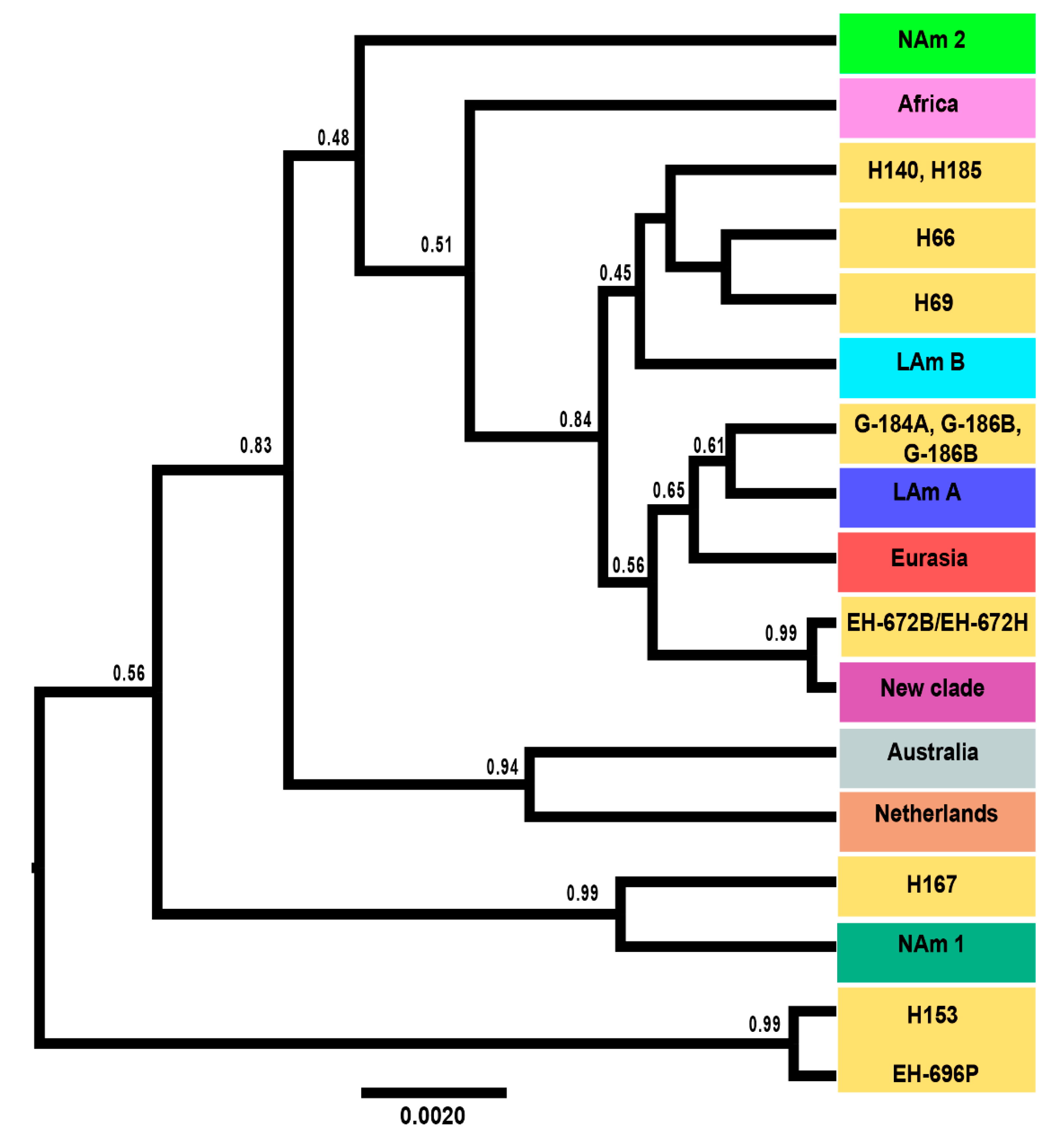
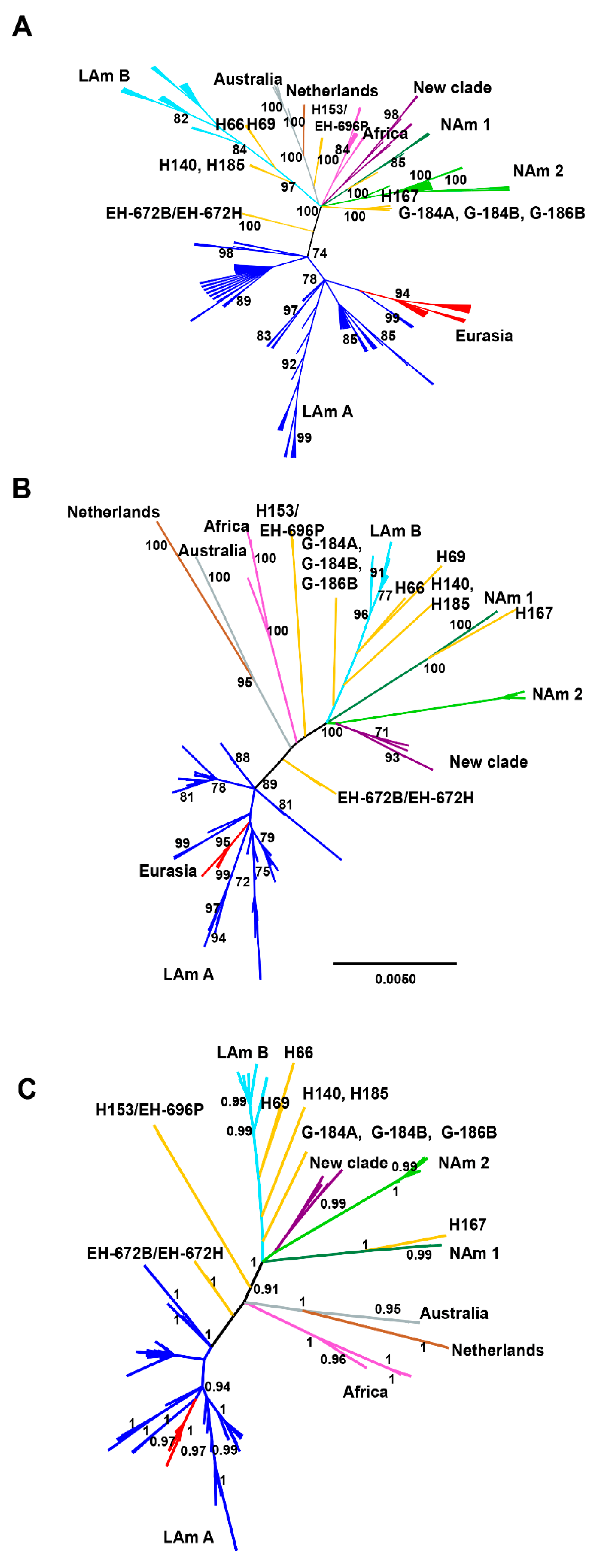
3.3. Phylogenetic Reconstruction
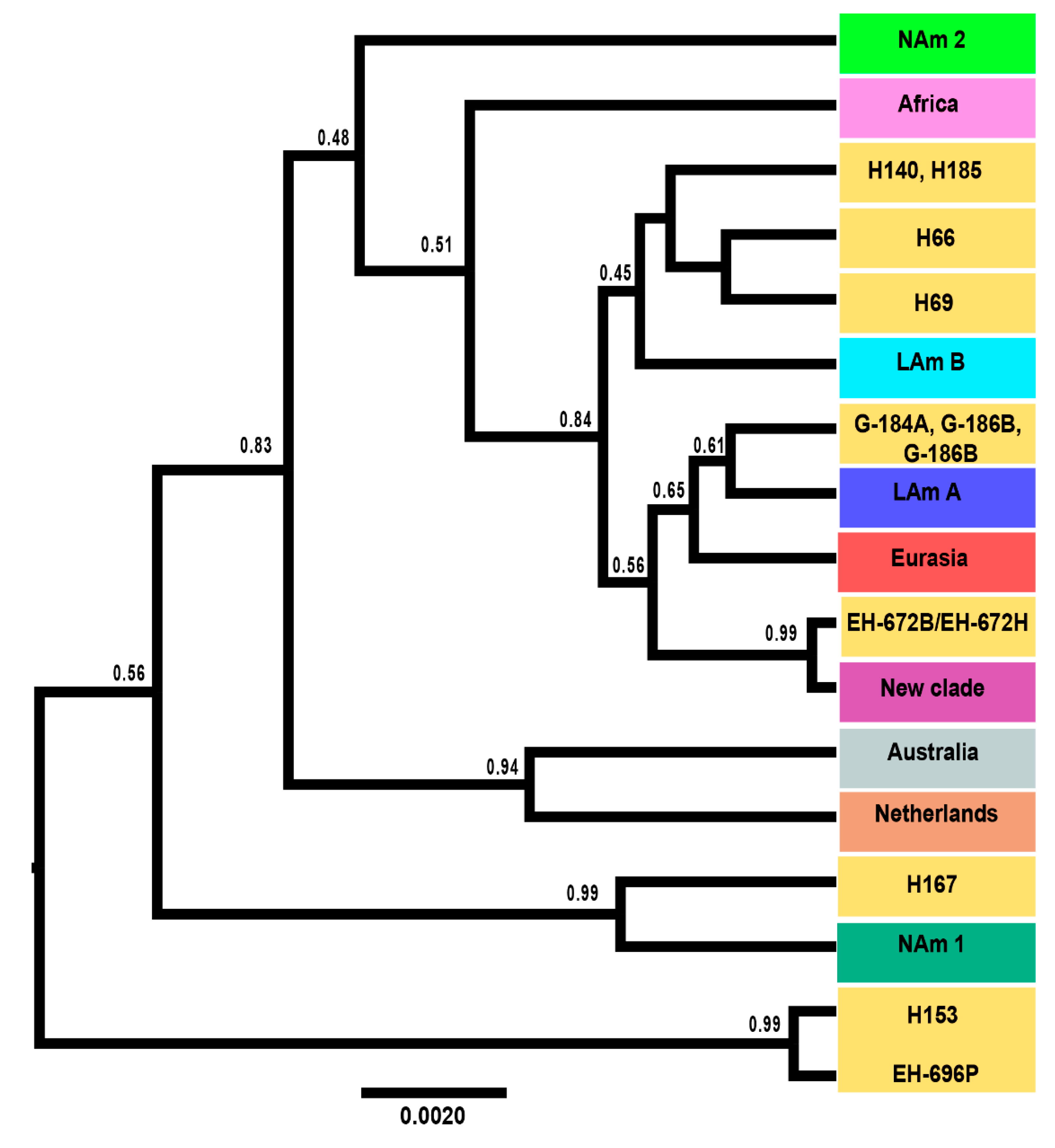
3.4. Coalescence Analysis
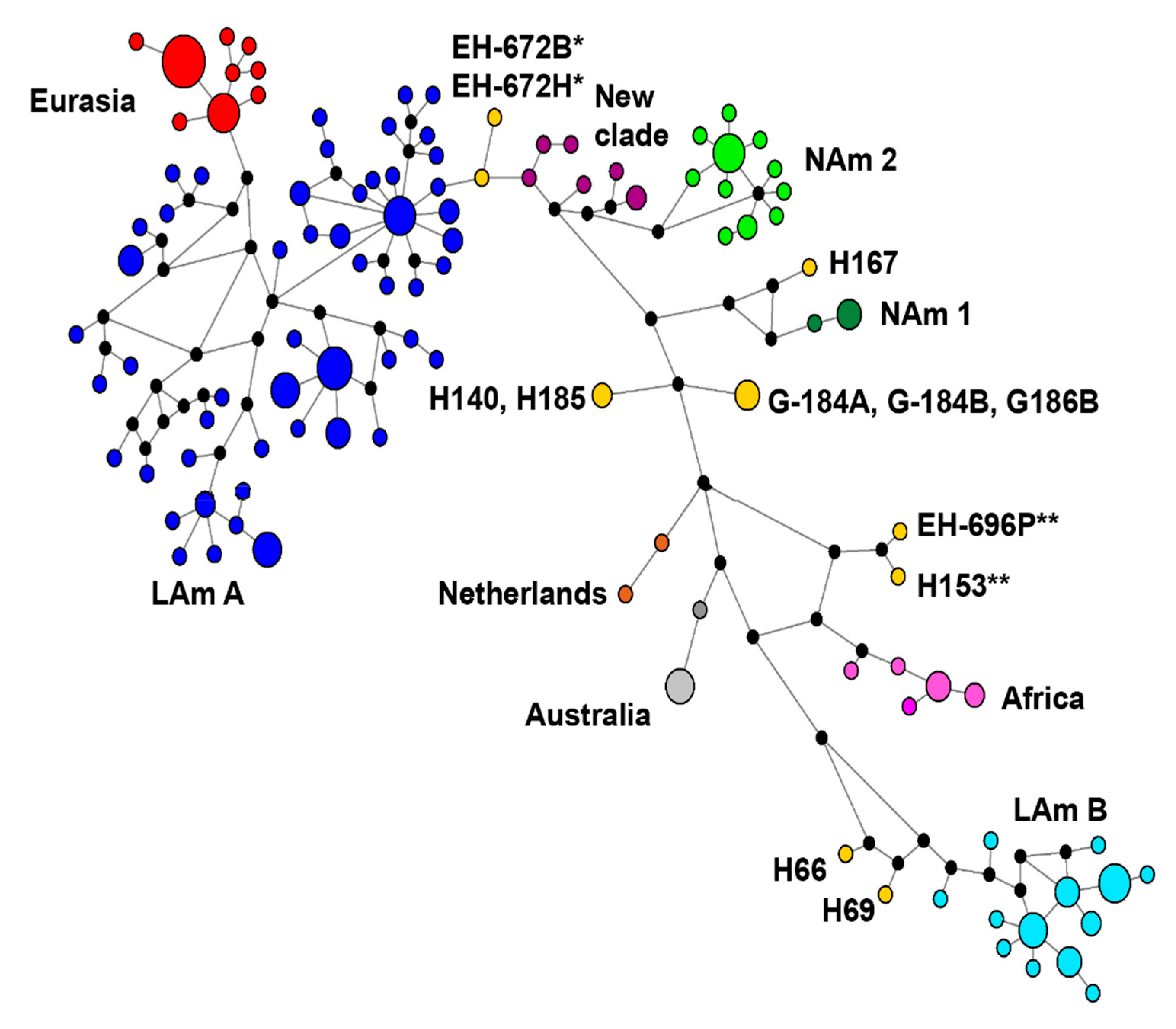
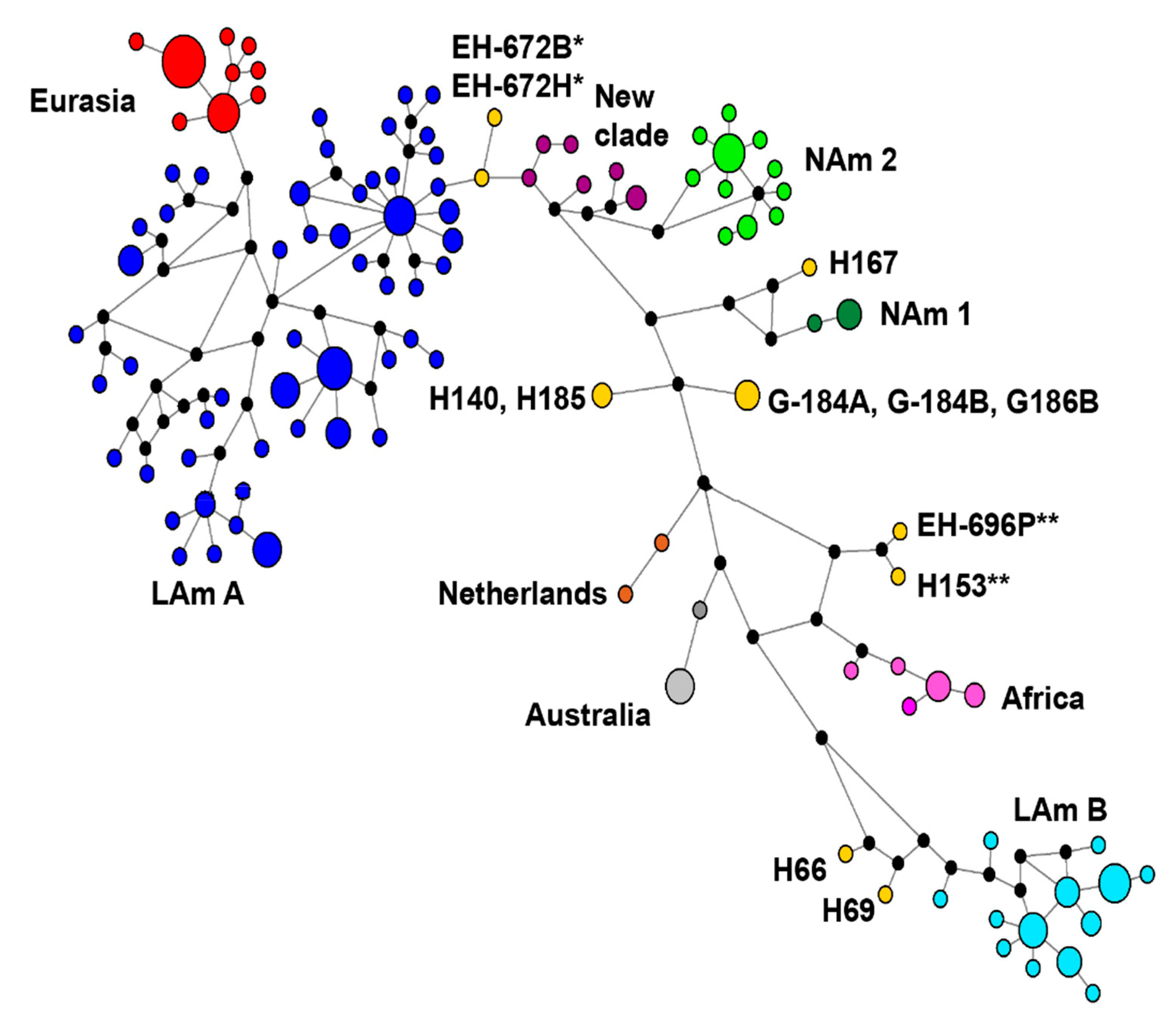
3.5. Concatenated Sequence-Types (CSTs) Network
3.6. Nucleotide Diversity (π)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, M.L.; Chávez-Tapia, C.B.; Vargas-Yañez, R.; Rodríguez-Arellanes, G.; Peña-Sandoval, G.R.; Toriello, C.; Pérez, A.; Reyes-Montes, M.R. Environmental conditions favoring bat infection with Histoplasma capsulatum in Mexican shelters. Am. J. Trop. Med. Hyg. 1999, 61, 914–919. [Google Scholar] [CrossRef] [Green Version]
- Canteros, C.E.; Iachini, R.H.; Rivas, M.C.; Vaccaro, O.; Madariaga, J.; Galarza, R.; Snaiderman, L.; Martínez, M.; Paladino, M.; Cicuttin, G.; et al. Primer aislamiento de Histoplasma capsulatum de murciélago urbano Eumops bonariensis. Rev. Argent. Microbiol. 2005, 37, 46–56. [Google Scholar] [PubMed]
- González-González, A.E.; Aliouat-Denis, C.M.; Carreto-Binaghi, L.E.; Ramírez, J.A.; Rodríguez-Arellanes, G.; Demanche, C.; Chabé, M.; Aliouat, E.M.; Dei-Cas, E.; Taylor, M.L. An Hcp100 gene fragment reveals Histoplasma capsulatum presence in lungs of Tadarida brasiliensis migratory bats. Epidemiol. Infect. 2012, 140, 1955–1963. [Google Scholar] [CrossRef] [Green Version]
- González-González, A.E.; Ramírez, J.A.; Aliouat-Denis, C.M.; Demanche, C.; Aliouat, E.M.; Dei-Cas, E.; Chabé, M.; Taylor, M.L. Molecular detection of Histoplasma capsulatum in the lung of a free-ranging common Noctule (Nyctalus noctula) from France using the Hcp100 gene. J. Zoo Wildl. Med. 2013, 44, 15–20. [Google Scholar] [CrossRef] [PubMed]
- González-González, A.E.; Aliouat-Denis, C.M.; Ramírez-Bárcenas, J.A.; Demanche, C.; Pottier, M.; Carreto-Binaghi, L.E.; Akbar, H.; Derouiche, S.; Chabé, M.; Aliouat, E.M.; et al. Histoplasma capsulatum and Pneumocystis spp. co-infection in wild bats from Argentina, French Guyana, and Mexico. BMC Microbiol. 2014, 14, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Alvarez, R.O.; Sahaza, J.H.; Berzunza-Cruz, M.; Becker, I.; Curiel-Quesada, E.; Pérez-Torres, A.; Reyes-Montes, M.R.; Taylor, M.L. Dimorphism and dissemination of Histoplasma capsulatum in the upper respiratory tract after intranasal infection of bats and mice with mycelial propagules. Am. J. Trop. Med. Hyg. 2019, 101, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, G.L.; Bigler, W.J. The role of bats in the propagation and spread of histoplasmosis: A review. J. Wildl. Dis. 1981, 17, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.L.; Chávez-Tapia, C.B.; Reyes-Montes, M.R. Molecular typing of Histoplasma capsulatum isolated from infected bats, captured in Mexico. Fungal Genet. Biol. 2000, 30, 207–212. [Google Scholar] [CrossRef]
- Taylor, M.L.; Hernández-García, L.; Estrada-Bárcenas, D.A.; Salas-Lizana, R.; Zancopé-Oliveira, R.M.; García de la Cruz, S.; Galvão-Dias, M.A.; Curiel-Quesada, E.; Canteros, C.E.; Bojórquez-Torres, G.; et al. Genetic diversity of microsatellite (GA)n and their flanking regions of Histoplasma capsulatum isolated from bats captured in three Latin-American countries. Fungal Biol. 2012, 116, 308–317. [Google Scholar] [CrossRef]
- Vincent, R.D.; Goewert, R.; Goldman, W.E.; Kobayashi, G.S.; Lambowitz, A.M.; Medoff, G. Classification of Histoplasma capsulatum isolates by restriction fragment polymorphisms. J. Bacteriol. 1986, 165, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Spitzer, E.D.; Lasker, B.A.; Travis, S.J.; Kobayashi, G.S.; Medoff, G. Use of mitochondrial and ribosomal DNA polymorphisms to classify clinical and soil isolates of Histoplasma capsulatum. Infect. Immun. 1989, 57, 1409–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitzer, E.D.; Keath, E.J.; Travis, S.J.; Painter, A.A.; Kobayashi, G.S.; Medoff, G. Temperature-sensitive variants of Histoplasma capsulatum isolated from patients with acquired immunodeficiency syndrome. J. Infect. Dis. 1990, 162, 258–261. [Google Scholar] [CrossRef]
- Keath, E.J.; Kobayashi, G.S.; Medoff, G. Typing of Histoplasma capsulatum by restriction fragment length polymorphisms in a nuclear gene. J. Clin. Microbiol. 1992, 30, 2104–2107. [Google Scholar] [CrossRef] [Green Version]
- Poonwan, N.; Imai, T.; Na, M.; Yazawa, K.; Mikami, Y.; Ando, A.; Nagata, Y. Genetic analysis of Histoplasma capsulatum strains isolated from clinical specimens in Thailand by a PCR-based random amplified polymorphic DNA method. J. Clin. Microbiol. 1998, 36, 3073–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Montes, M.R.; Bobadilla-Del Valle, M.; Martínez-Rivera, M.A.; Rodríguez-Arellanes, G.; Maravilla, E.; Sifuentes-Osornio, J.; Taylor, M.L. Relatedness analyses of Histoplasma capsulatum isolates from Mexican patients with AIDS-associated histoplasmosis by using histoplasmin electrophoretic profiles and randomly amplified polymorphic DNA patterns. J. Clin. Microbiol. 1999, 37, 1404–1408. [Google Scholar] [CrossRef] [Green Version]
- Muniz, M. de M.; Pizzini, C.V.; Peralta, J.M.; Reiss, E.; Zancopé-Oliveira, R.M. Genetic diversity of Histoplasma capsulatum strains isolated from soil, animals, and clinical specimens in Rio de Janeiro State, Brazil, by a PCR-based random amplified polymorphic DNA assay. J. Clin. Microbiol. 2001, 39, 4487–4494. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, T.; Taylor, J.W.; White, T.J. Phylogenetic relationships of varieties and geographical groups of the human pathogenic fungus Histoplasma capsulatum Darling. J. Clin. Microbiol. 1999, 37, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Bartlett, M.; Allen, S.D.; Smith, J.W.; Wheat, L.J.; Connolly, P.A.; Lee, C.H. Typing of Histoplasma capsulatum isolates based on nucleotide sequence variation in the Internal Transcribed Spacer regions of rRNA genes. J. Clin. Microbiol. 2000, 38, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.A.; Taylor, J.W.; Dechairo, B.; Burt, A.; Koenig, G.L.; White, T.J. Amplified single-nucleotide polymorphisms and a (GA)n microsatellite marker reveal genetic differentiation between populations of Histoplasma capsulatum from the Americas. Fungal Genet. Biol. 2001, 34, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, T.; White, T.J.; Koenig, G.; McEwen, J.; Restrepo, A.; Castañeda, E.; Da Silva Lacaz, C.; Heins-Vaccari, E.M.; De Freitas, R.S.; Zancopé-Oliveira, R.M.; et al. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 2003, 12, 3383–3401. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.L.; Chávez-Tapia, C.B.; Rojas-Martínez, A.; Reyes-Montes, M.R.; Bobadilla-Del Valle, M.; Zúñiga, G. Geographical distribution of genetic polymorphism of the pathogen Histoplasma capsulatum isolated from infected bats, captured in a central zone of Mexico. FEMS Immunol. Med. Microbiol. 2005, 45, 451–458. [Google Scholar] [CrossRef] [Green Version]
- De Muniz, M.M.; Morais, P.M.S.; Meyer, W.; Nosanchuk, J.D.; Zancopé-Oliveira, R.M. Comparison of different DNA-based methods for molecular typing of Histoplasma capsulatum. Appl. Environ. Microbiol. 2010, 76, 4438–4447. [Google Scholar] [CrossRef] [Green Version]
- Balajee, S.A.; Hurst, S.F.; Chang, L.S.; Miles, M.; Beeler, W.; Hale, C.; Kasuga, T.; Benedict, K.; Chiller, T.; Lindsley, M.D. Multilocus sequence typing of Histoplasma capsulatum in formalin-fixed paraffin-embedded tissues from cats living in non-endemic regions reveals a new phylogenetic clade. Med. Mycol. 2013, 51, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Galo, C.; Sanchez, A.L.; Fontecha, G.A. Genetic diversity of Histoplasma capsulatum isolates from Honduras. Sci. J. Microbiol. 2013. [Google Scholar] [CrossRef]
- Vite-Garín, T.; Estrada-Bárcenas, D.A.; Cifuentes, J.; Taylor, M.L. The importance of molecular analyses for understanding the genetic diversity of Histoplasma capsulatum: An overview. Rev. Iberoam. Micol. 2014, 31, 11–15. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Patané, J.S.L.; Taylor, M.L.; Gómez, B.L.; Theodoro, R.C.; de Hoog, S.; Engelthaler, D.M.; Zancopé-Oliveira, R.M.; Felipe, M.S.S.; Barker, B.M. Worldwide phylogenetic distributions and population dynamics of the genus Histoplasma. PLoS Negl. Trop. Dis. 2016, 10, e0004732. [Google Scholar] [CrossRef] [Green Version]
- Sepúlveda, V.E.; Márquez, R.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Genome sequences reveal cryptic speciation in the human pathogen Histoplasma capsulatum. MBio 2017, 8, e01339-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, C.S.; Sepúlveda, V.E.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Recent admixture between species of the fungal pathogen Histoplasma. Evol. Let. 2018, 2, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibayrenc, M. Towards a unified evolutionary genetics of microorganisms. Annu. Rev. Microbiol. 1996, 50, 401–429. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arellanes, G.; Pérez-Mejía, A.; Duarte-Escalante, E.; Taylor, M.L. Organización de la colección de cepas de Histoplasma capsulatum del Laboratorio de Inmunología de Hongos, Facultad de Medicina, UNAM. Rev. Inst. Nal. Enf. Resp. Mex. 1998, 11, 243–246. [Google Scholar]
- Gannon, W.L.; Sikes, R.S. The Animal Care and Use Committee of the American Society of Mammalogists: Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal. 2007, 88, 809–823. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Farris, J.S.; Källersjö, M.; Kluge, A.G.; Bult, C. Testing significance of incongruence. Cladistics 1994, 10, 315–319. [Google Scholar] [CrossRef]
- Goloboff, P.A.; Farris, J.S.; Nixon, K.C. TNT, a free program for phylogenetic analysis. Cladistics 2008, 24, 774–786. [Google Scholar] [CrossRef]
- Nixon, K.C. The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 1999, 15, 407–414. [Google Scholar] [CrossRef]
- Silvestro, D.; Michalak, I. RaxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohnä, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model selection across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. JModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Heled, J.; Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayden, R.L. A hierarchy of species concepts: The denouement in the saga of the species problem. In Species. The Units of Biodiversity; Oaridge, M.F., Dawah, H.A., Wilson, M.R., Eds.; Chapman and Hall: London, UK, 1997; pp. 381–424. [Google Scholar]
- Arita, H.T.; Ortega, J. Tadarida brasiliensis (I. Geoffroy, 1824). In Los Mamíferos Silvestres de México; Ceballos, G., Oliva, G., Eds.; Fondo de Cultura Económica/CONABIO: Ciudad de México, MX, USA, 2005; pp. 335–337. [Google Scholar]
- Iñiguez-Dávalos, L.I. Mormoops megalophylla (Peters, 1864). In Los Mamíferos Silvestres de México; Ceballos, G., Oliva, G., Eds.; Fondo de Cultura Económica/CONABIO: Ciudad de México, MX, USA, 2005; pp. 178–179. [Google Scholar]
- Russel, A.L.; Medellin, R.A.; McCracken, G.F. Genetic variation and migration in the Mexican free-tailed bat Tadarida brasiliensis mexicana. Mol. Ecol. 2005, 14, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Hipp, A.L.; Hall, J.C.; Sytsma, K.J. Congruence versus phylogenetic accuracy: Revisiting the incongruence length difference test. Syst. Biol. 2004, 53, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sites, J.W., Jr.; Marshall, J.C. Operational criteria for delimiting species. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 199–227. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Related | Phylogenetic- | Origin | GenBank (Accession Numbers) | ||||
|---|---|---|---|---|---|---|---|
| Acronym | Source | Species/Lineage a | arf | H-anti | ole1 | tub1 | |
| 1558 | Human | LAm B (*) | AR | KT601344 | KT601418 | KT601381 | KT601463 |
| 1739 | Human | LAm B (*) | AR | KT601345 | KT601419 | KT601382 | KT601464 |
| 92590 | Human | LAm B (LAm B1) | AR | KT601346 | KT601420 | KT601383 | KT601465 |
| 951814 | Human | LAm B (LAm B1) | AR | KT601347 | KT601421 | KT601384 | KT601466 |
| 993444 | Human | LAm B (LAm B1) | AR | KT601348 | KT601423 | KT601385 | KT601467 |
| 993445 | Human | LAm B (LAm B1) | AR | KT601349 | KT601424 | KT601386 | KT601468 |
| 993446 | Human | LAm B (LAm B1) | AR | KT601350 | KT601425 | KT601387 | KT601469 |
| 993267 | Human | LAm B (LAm B1) | AR | KT601351 | KT601422 | KT601388 | KT601470 |
| AP | Human | LAm A (LAm A1) | CO | KT601352 | KT601427 | KT601389 | KT601471 |
| DS | Human | LAm A (*) | CO | KT601353 | KT601428 | KT601390 | KT601472 |
| GeM | Human | LAm A (LAm A1) | CO | KT601354 | KT601445 | KT601391 | KT601473 |
| GLi | Human | LAm A (LAm A2) | CO | KT601355 | KT601446 | KT601392 | KT601474 |
| Hz | Human | LAm B (LAm B1) | CO | KT601356 | KT601449 | KT601393 | KT601475 |
| JG | Human | LAm B (LAm B1) | CO | KT601357 | KT601450 | KT601394 | KT601476 |
| LA | Human | LAm A (LAm A2) | CO | KT601358 | KT601451 | KT601395 | KT601477 |
| LF | Human | LAm A (*) | CO | KT601359 | KT601452 | KT601396 | KT601478 |
| MZ2 | Human | LAm A (LAm A2) | CO | KT601360 | KT601426 | KT601397 | KT601479 |
| RG | Human | LAm A (LAm A1) | CO | KT601361 | KT601453 | KT601398 | KT601480 |
| WCh | Human | LAm A (LAm A1) | CO | KT601362 | KT601454 | KT601399 | KT601481 |
| H.1.02.W | Human | LAm A (LAm A2) | GT | KT601363 | KT601447 | KT601400 | KT601482 |
| H.1.12.96 | Human | LAm A (LAm A2) | GT | KT601364 | KT601448 | KT601401 | KT601483 |
| EH-323 | Human | LAm A (*) | MX | KT601365 | KT601429 | KT601402 | KT601484 |
| EH-324 | Human | LAm A (*) | MX | KT601366 | KT601430 | KT601403 | KT601485 |
| EH-326 | Human | LAm A (*) | MX | KT601367 | KT601431 | KT601404 | KT601486 |
| EH-327 | Human | LAm A (*) | MX | KT601368 | KT601432 | KT601405 | KT601487 |
| EH-328 | Human | LAm A (LAm A1) | MX | KT601369 | KT601433 | KT601406 | KT601488 |
| EH-355 | Human | LAm A (*) | MX | KT601370 | KT601434 | KT601407 | KT601489 |
| EH-356 | Human | LAm A (*) | MX | KT601371 | KT601435 | KT601408 | KT601490 |
| EH-357 | Human | LAm A (*) | MX | KT601372 | KT601436 | KT601409 | KT601491 |
| EH-383I b | L. nivalis | LAm A (LAm A1) | MX | AF495619 | AF495620 | AF495621 | F495622 |
| EH-383P b | L. nivalis | LAm A (LAm A1) | MX | AF495623 | AF495624 | AF495625 | AF495626 |
| EH-384I b | T. brasiliensis | NAm 3 (BAC1) | MX | AF495627 | AF495628 | AF495629 | AF495630 |
| EH-384P b | T. brasiliensis | NAm 3 (BAC1) | MX | AF495631 | AF495632 | AF495633 | AF495634 |
| EH-408H b | L. nivalis | LAm A (LAm A1) | MX | AF495644 | AF495643 | AF495645 | AF495646 |
| EH-449B | L. nivalis | LAm A (LAm A1 | MX | KT601373 | KT601437 | KT601410 | KT601455 |
| EH-655P | T. brasiliensis | NAm 3 (BAC1) | MX | KT601374 | KT601438 | KT601411 | KT601458 |
| EH-658H | T. brasiliensis | NAm 3 (BAC1) | MX | KT601375 | KT601439 | KT601412 | KT601459 |
| EH-670B | T. brasiliensis | NAm 3 (BAC1) | MX | KT601376 | KT601440 | KT601414 | KT601460 |
| EH-670H | T. brasiliensis | NAm 3 (BAC1) | MX | KT601377 | KT601441 | KT601415 | KT601461 |
| EH-672B | T. brasiliensis | NAm 3 (*) | MX | KT601378 | KT601442 | KT601413 | KT601456 |
| EH-672H | T. brasiliensis | NAm 3 (*) | MX | KT601379 | KT601443 | KT601416 | KT601457 |
| EH-696P | T. brasiliensis | H153-lineage (*) | MX | KT601380 | KT601444 | KT601417 | KT601462 |
| Gene Fragments | Nucleotide Sites | ||||
|---|---|---|---|---|---|
| Size (nt) | a Start/End (nt) | Variable | Parsimonious | ||
| Informative | Uninformative | ||||
| arf | 457 | 415/871 | 70 | 44 | 26 |
| H-anti | 397 | 394/789 | 91 | 59 | 32 |
| ole1 | 414 | 37/450 | 69 | 49 | 20 |
| tub1 | 270 | 590/862 | 91 | 74 | 17 |
| Total | 1538 | 321 | 226 | 95 | |
| Sum of Tree Lengths | |||
|---|---|---|---|
| Partition | Original Partition | Range of Replicates | p Value |
| Four genes | 1243 | 1243–1242 | 0.997 |
| arf vs. H-anti | 161 | 161–0 | 1 |
| arf vs. ole1 | 156 | 156–157 | 0.941 |
| arf vs. tub1 | 193 | 193–194 | 0.997 |
| H-anti vs. ole1 | 168 | 168–169 | 0.748 |
| H-anti vs. tub1 | 217 | 217–0 | 1 |
| ole1 vs. tub1 | 202 | 202–0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vite-Garín, T.; Estrada-Bárcenas, D.A.; Gernandt, D.S.; Reyes-Montes, M.d.R.; Sahaza, J.H.; Canteros, C.E.; Ramírez, J.A.; Rodríguez-Arellanes, G.; Serra-Damasceno, L.; Zancopé-Oliveira, R.M.; et al. Histoplasma capsulatum Isolated from Tadarida brasiliensis Bats Captured in Mexico Form a Sister Group to North American Class 2 Clade. J. Fungi 2021, 7, 529. https://doi.org/10.3390/jof7070529
Vite-Garín T, Estrada-Bárcenas DA, Gernandt DS, Reyes-Montes MdR, Sahaza JH, Canteros CE, Ramírez JA, Rodríguez-Arellanes G, Serra-Damasceno L, Zancopé-Oliveira RM, et al. Histoplasma capsulatum Isolated from Tadarida brasiliensis Bats Captured in Mexico Form a Sister Group to North American Class 2 Clade. Journal of Fungi. 2021; 7(7):529. https://doi.org/10.3390/jof7070529
Chicago/Turabian StyleVite-Garín, Tania, Daniel A. Estrada-Bárcenas, David S. Gernandt, María del Rocío Reyes-Montes, Jorge H. Sahaza, Cristina E. Canteros, José A. Ramírez, Gabriela Rodríguez-Arellanes, Lisandra Serra-Damasceno, Rosely M. Zancopé-Oliveira, and et al. 2021. "Histoplasma capsulatum Isolated from Tadarida brasiliensis Bats Captured in Mexico Form a Sister Group to North American Class 2 Clade" Journal of Fungi 7, no. 7: 529. https://doi.org/10.3390/jof7070529