
Simplified All-In-One CRISPR-Cas9 Construction for Efficient Genome Editing in Cryptococcus Species


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Media and Reagents
2.2. Construction of pRH003 and pNK003 Plasmids
2.3. Cloning of the Target Sequence into the gRNA Cassette
2.4. Introduction of Donor DNA with the In-Fusion Cloning Technique
2.5. Electroporation of Cryptococcus Species
2.6. Southern Blot Analysis
3. Results
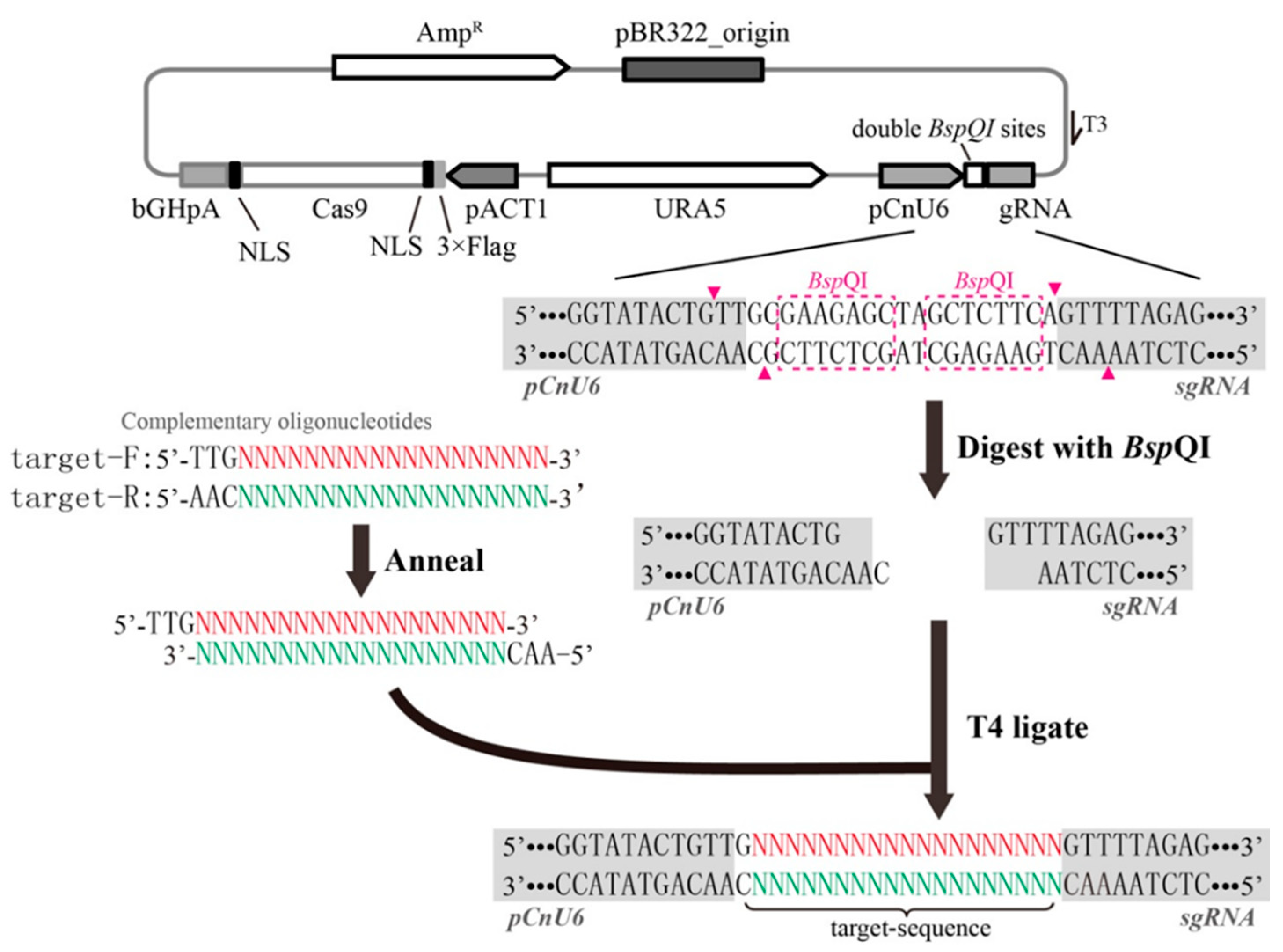
3.1. Rapid Cloning of the Target Sequence into the All-In-One CRISPR-Cas9 Vector
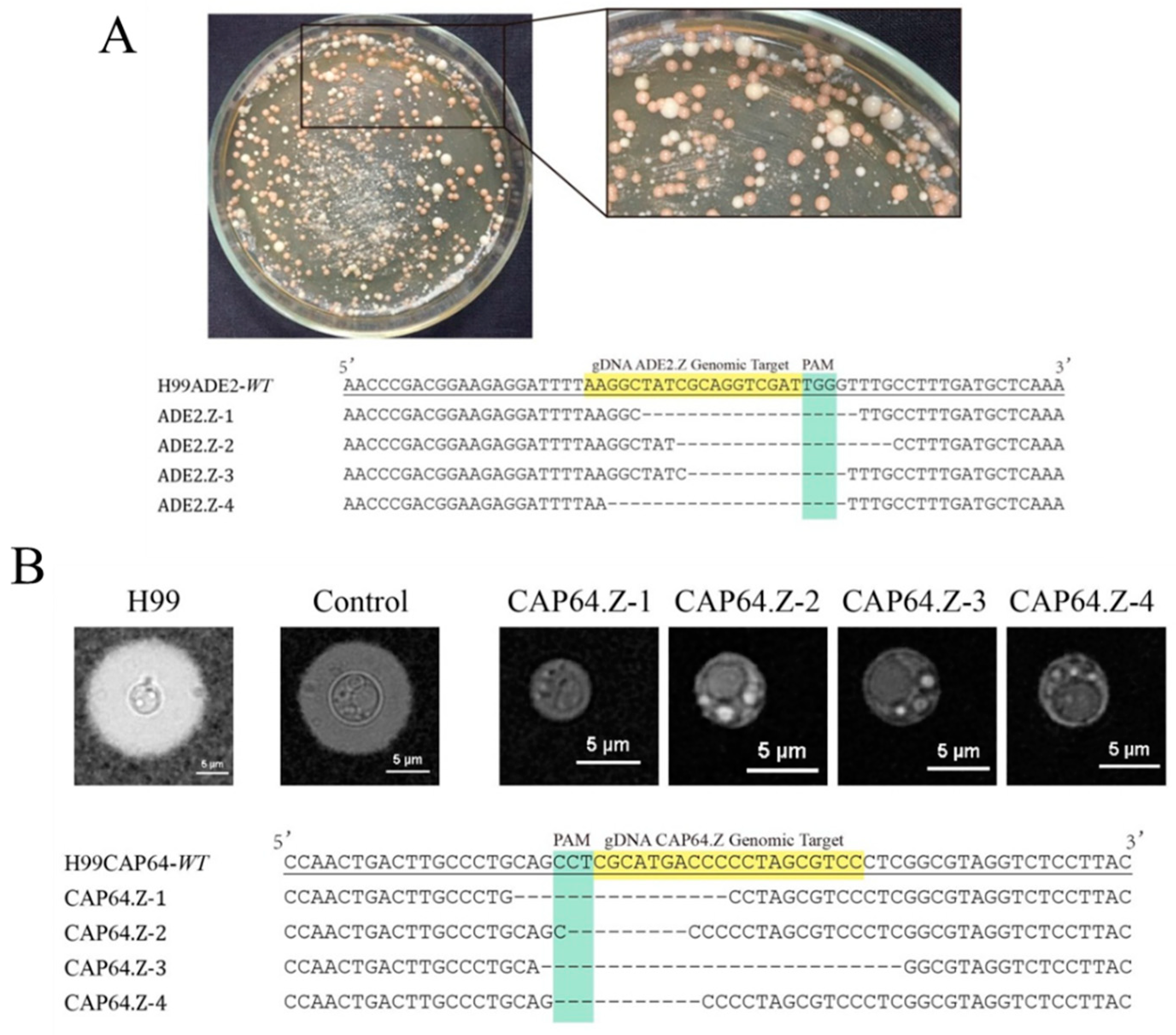
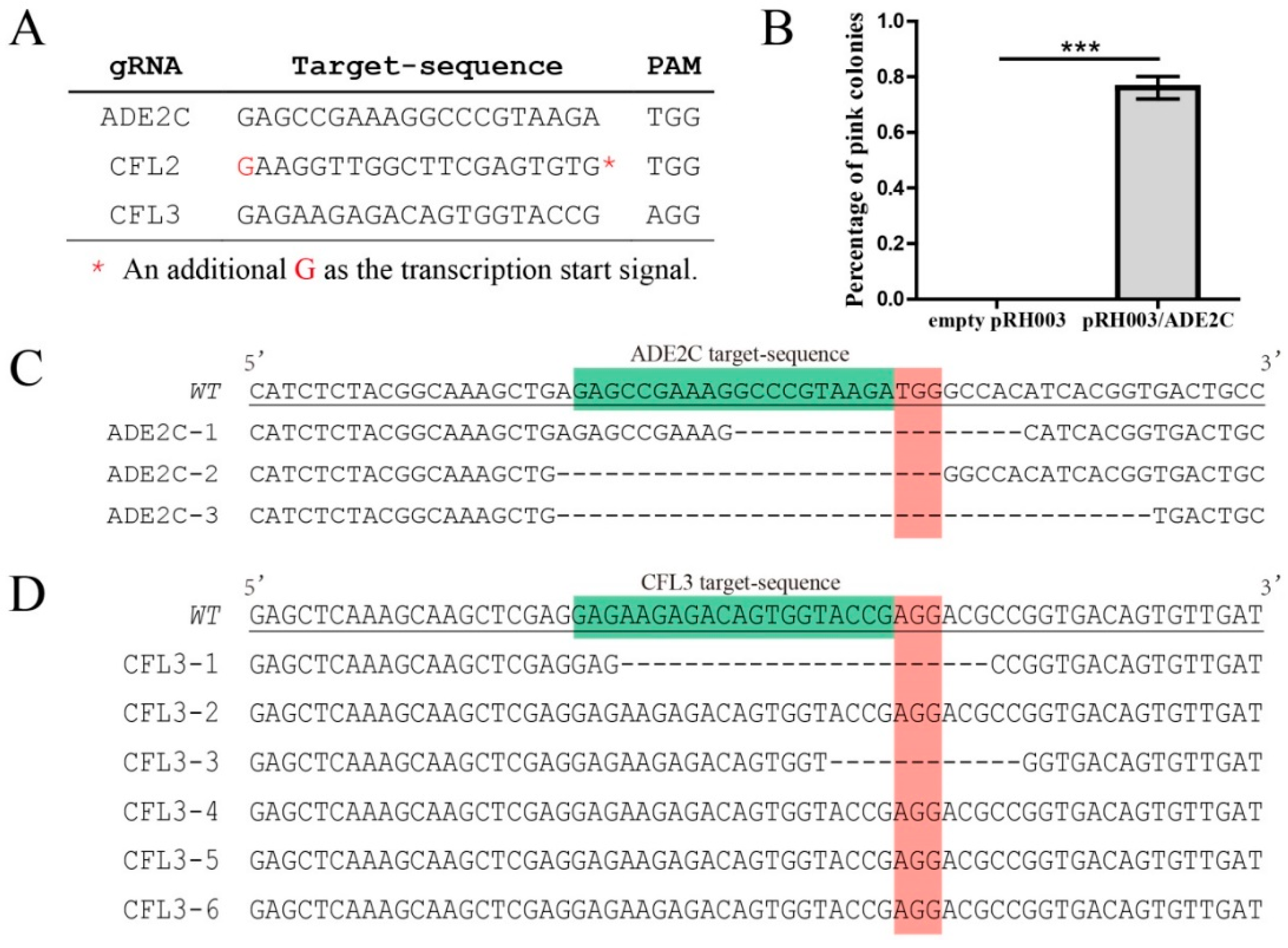
3.2. High Efficiency of the Simplified All-In-One Vector in Gene Mutagenesis
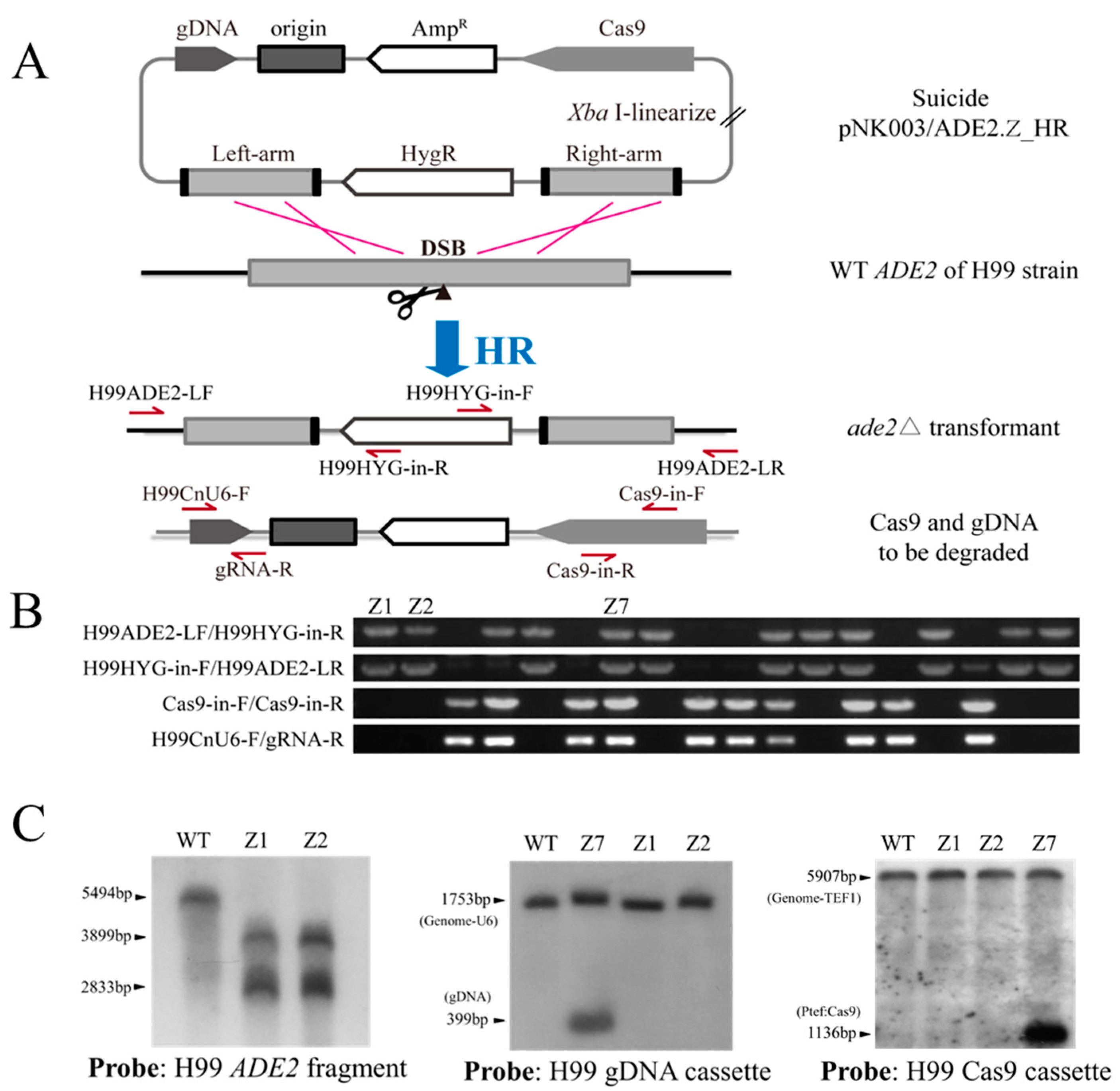
3.3. Knockout of Genes in One Step by the Simplified ‘Suicide’ CRISPR-Cas9 Vector
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wormley, F.L.; Perfect, J.R. Immunology of infection caused by Cryptococcus neoformans. Methods Mol. Med. 2005, 118, 193–198. [Google Scholar] [PubMed]
- Idnurm, A.; Bahn, Y.-S.; Nielsen, K.; Lin, X.; Fraser, J.A.; Heitman, J. Deciphering the model pathogenic fungus Cryptococcus neoformans. Nat. Rev. Microbiol. 2005, 3, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R. Cryptococcosis: A model for the understanding of infectious diseases. J. Clin. Investig. 2014, 124, 1893–1895. [Google Scholar] [CrossRef] [Green Version]
- Edman, J.C.; Kwon-Chung, K. Isolation of the URA5 gene from Cryptococcus neoformans var. neoformans and its use as a selective marker for transformation. Mol. Cell. Biol. 1990, 10, 4538–4544. [Google Scholar]
- Toffaletti, D.L.; Rude, T.H.; Johnston, S.A.; Durack, D.; Perfect, J. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 1993, 175, 1405–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, R.C.; Cruz, M.C.; Sia, R.A.; Allen, B.; Alspaugh, J.A.; Heitman, J. Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans. Fungal Genet. Biol. 2000, 29, 38–48. [Google Scholar] [CrossRef]
- Chang, Y.; Kwon-Chung, K. Isolation of the third capsule-associated gene, CAP60, required for virulence in Cryptococcus neoformans. Infect. Immun. 1998, 66, 2230–2236. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Kwon-Chung, K. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol. Cell. Biol. 1994, 14, 4912–4919. [Google Scholar] [CrossRef]
- Chang, Y.C.; Penoyer, L.A.; Kwon-Chung, K. The second capsule gene of Cryptococcus neoformans, CAP64, is essential for virulence. Infect. Immun. 1996, 64, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Salas, S.; Bennett, J.; Kwon-Chung, K.; Perfect, J.; Williamson, P. Effect of the laccase gene CNLAC1, on virulence of Cryptococcus neoformans. J. Exp. Med. 1996, 184, 377–386. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Chen, L.; Jiang, Y.; Zhou, Z.; Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov. 2015, 1, 15007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, C.M.; Hussain, M.S.; Blenner, M.; Wheeldon, I. Synthetic RNA polymerase III promoters facilitate high-efficiency CRISPR–Cas9-mediated genome editing in Yarrowia lipolytica. ACS Synth. Biol. 2016, 5, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.C.; Platt, R.J.; Goldfless, S.J.; Zhang, F.; Niles, J.C. Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat. Methods 2014, 11, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Morio, F.; Lombardi, L.; Butler, G. The CRISPR toolbox in medical mycology: State of the art and perspectives. PLoS Pathog. 2020, 16, e1008201. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wei, D.; Zhu, X.; Pan, J.; Zhang, P.; Huo, L.; Zhu, X. A ‘suicide’ CRISPR-Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci. Rep. 2016, 6, 31145. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Lin, X. Multiple applications of a transient CRISPR-Cas9 coupled with electroporation (TRACE) system in the Cryptococcus neoformans species complex. Genetics 2018, 208, 1357–1372. [Google Scholar] [CrossRef] [Green Version]
- Wang, P. Two distinct approaches for CRISPR-Cas9-mediated gene editing in Cryptococcus neoformans and related species. Msphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.Z.; Ciccaglione, K.M.; Tournier, V.; Zaratiegui, M. Implementation of the CRISPR-Cas9 system in fission yeast. Nat. Commun. 2014, 5, 5344. [Google Scholar] [CrossRef]
- Arras, S.D.; Chua, S.M.; Wizrah, M.S.; Faint, J.A.; Yap, A.S.; Fraser, J.A. Targeted genome editing via CRISPR in the pathogen Cryptococcus neoformans. PLoS ONE 2016, 11, e0164322. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, L.E.; Rada, H.; Flanagan, J.; Diprose, J.M.; Gilbert, R.J.; Owens, R.J. Application of In-Fusion™ cloning for the parallel construction of E. coli expression vectors. Methods Mol. Biol. 2014, 1116, 209–234. [Google Scholar]
- Mansour, S.L.; Thomas, K.R.; Capecchi, M.R. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature 1988, 336, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Shen, B.; Zhang, C.; Huang, X.; Zhang, Y. sgRNAcas9: A software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 2014, 9, e100448. [Google Scholar] [CrossRef]
- Haber, J.E. Partners and pathways: Repairing a double-strand break. Trends Genet. 2000, 16, 259–264. [Google Scholar] [CrossRef]
- Li, Z.; Bi, J.; Yang, J.; Pan, J.; Sun, Z.; Zhu, X. Requirement of a Tsp2-type tetraspanin for laccase repression and stress resistance in the basidiomycete Cryptococcus neoformans. Appl. Environ. Microbiol. 2012, 78, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querin, L.; Sanvito, R.; Magni, F.; Busti, S.; Van Dorsselaer, A.; Alberghina, L.; Vanoni, M. Proteomic analysis of a nutritional shift-up in Saccharomyces cerevisiae identifies Gvp36 as a BAR-containing protein involved in vesicular traffic and nutritional adaptation. J. Biol. Chem. 2008, 283, 4730–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Cottrell, T.R.; Pierini, L.M.; Goldman, W.E.; Doering, T.L. RNA interference in the pathogenic fungus Cryptococcus neoformans. Genetics 2002, 160, 463–470. [Google Scholar] [CrossRef]
- Ost, K.S.; O’Meara, T.R.; Huda, N.; Esher, S.K.; Alspaugh, J.A. The Cryptococcus neoformans alkaline response pathway: Identification of a novel Rim pathway activator. PLoS Genet. 2015, 11, e1005159. [Google Scholar] [CrossRef] [Green Version]
- Matsu-Ura, T.; Baek, M.; Kwon, J.; Hong, C. Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol. Biotechnol. 2015, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nødvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar]
- Enkler, L.; Richer, D.; Marchand, A.L.; Ferrandon, D.; Jossinet, F. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci. Rep. 2016, 6, 35766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michielse, C.B.; Hooykaas, P.J.; van den Hondel, C.A.; Ram, A.F. Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr. Genet. 2005, 48, 1–17. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Wang, Y.; Li, C.; Ma, X.; Ma, L.; Zhu, X. Simplified All-In-One CRISPR-Cas9 Construction for Efficient Genome Editing in Cryptococcus Species. J. Fungi 2021, 7, 505. https://doi.org/10.3390/jof7070505
Zhang P, Wang Y, Li C, Ma X, Ma L, Zhu X. Simplified All-In-One CRISPR-Cas9 Construction for Efficient Genome Editing in Cryptococcus Species. Journal of Fungi. 2021; 7(7):505. https://doi.org/10.3390/jof7070505
Chicago/Turabian StyleZhang, Ping, Yu Wang, Chenxi Li, Xiaoyu Ma, Lan Ma, and Xudong Zhu. 2021. "Simplified All-In-One CRISPR-Cas9 Construction for Efficient Genome Editing in Cryptococcus Species" Journal of Fungi 7, no. 7: 505. https://doi.org/10.3390/jof7070505
APA StyleZhang, P., Wang, Y., Li, C., Ma, X., Ma, L., & Zhu, X. (2021). Simplified All-In-One CRISPR-Cas9 Construction for Efficient Genome Editing in Cryptococcus Species. Journal of Fungi, 7(7), 505. https://doi.org/10.3390/jof7070505