
Genotype, Antifungal Susceptibility, and Virulence of Clinical South African Cryptococcus neoformans Strains from National Surveillance, 2005–2009


 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Selection
2.2. Subculture and Identification of Surveillance Isolates
2.3. Multi Locus Sequence typing (MLST) Experiments and Data Analysis
2.4. Fluconazole Susceptibility Testing
2.5. Virulence Studies
2.6. Whole Genome Sequencing and Phylogenetic Analysis
2.7. Statistical Analyses
2.8. Ethics Approval
3. Results
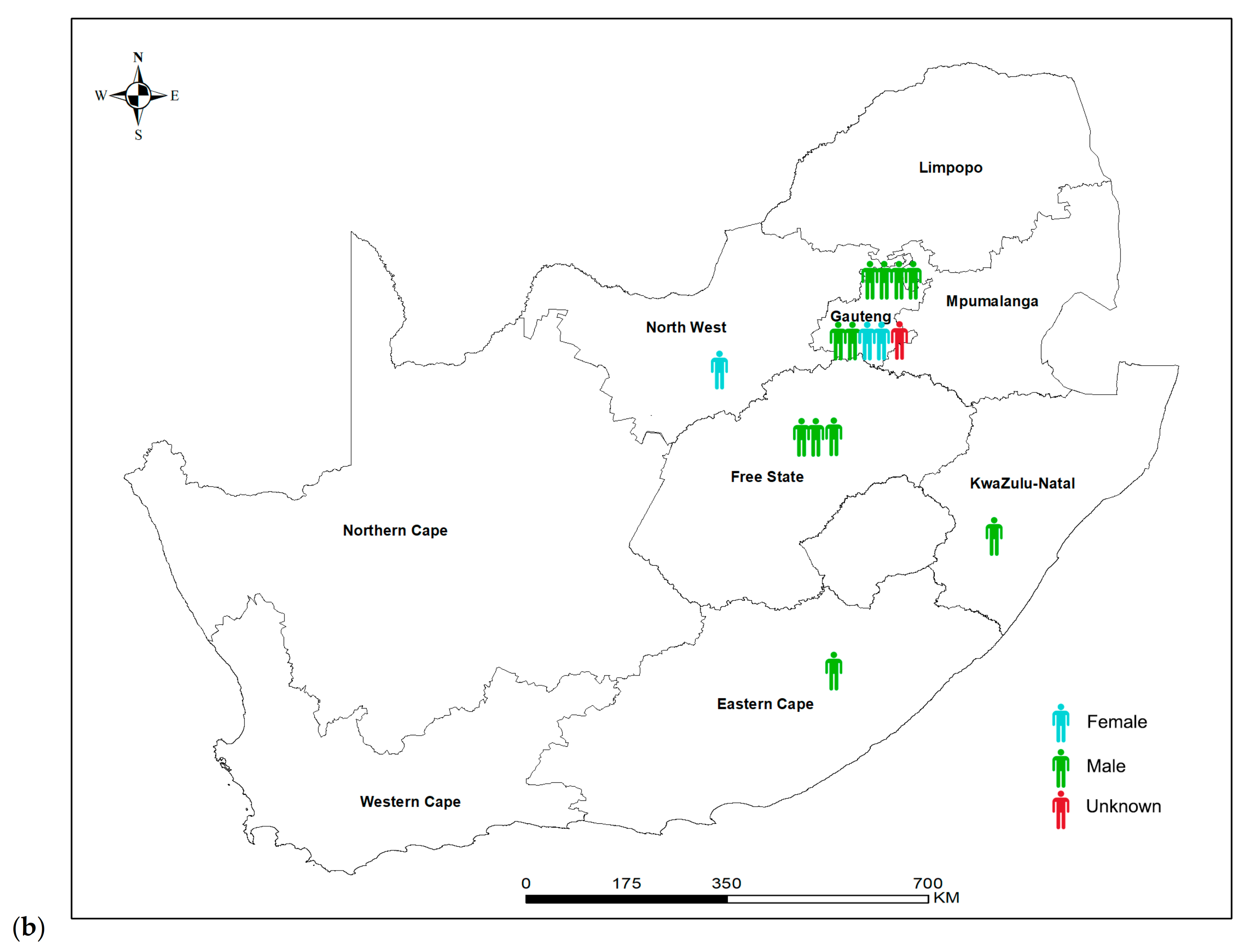
3.1. Descriptive Analysis of National Laboratory-Based Surveillance
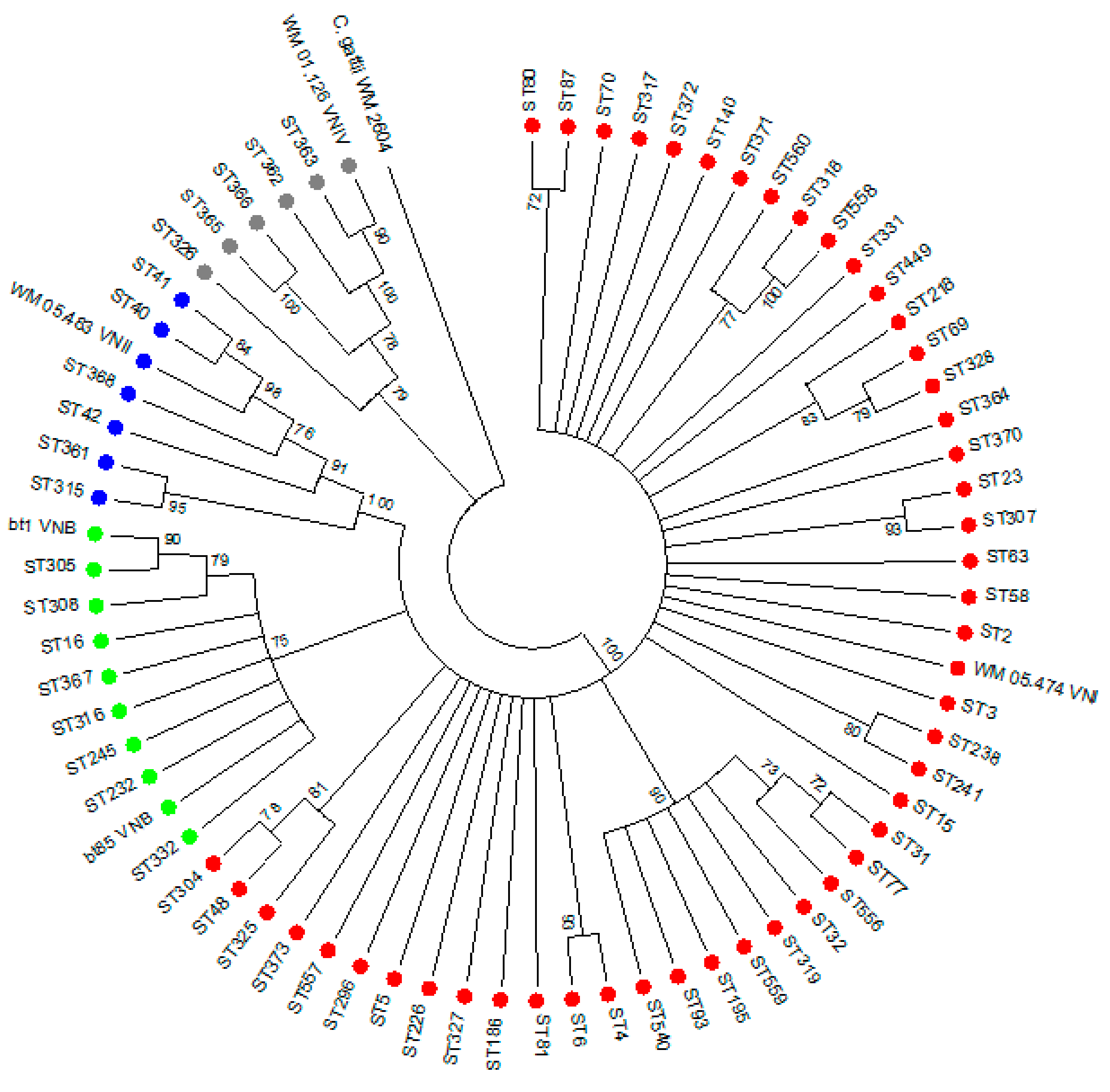
3.2. Genotyping of 251 Clinical C. neoformans Isolates
3.3. Association between Patients’ Clinical Characteristics and Genotype
3.4. Association between Genotype and In-Hospital Mortality
3.5. Fluconazole Susceptibility Testing
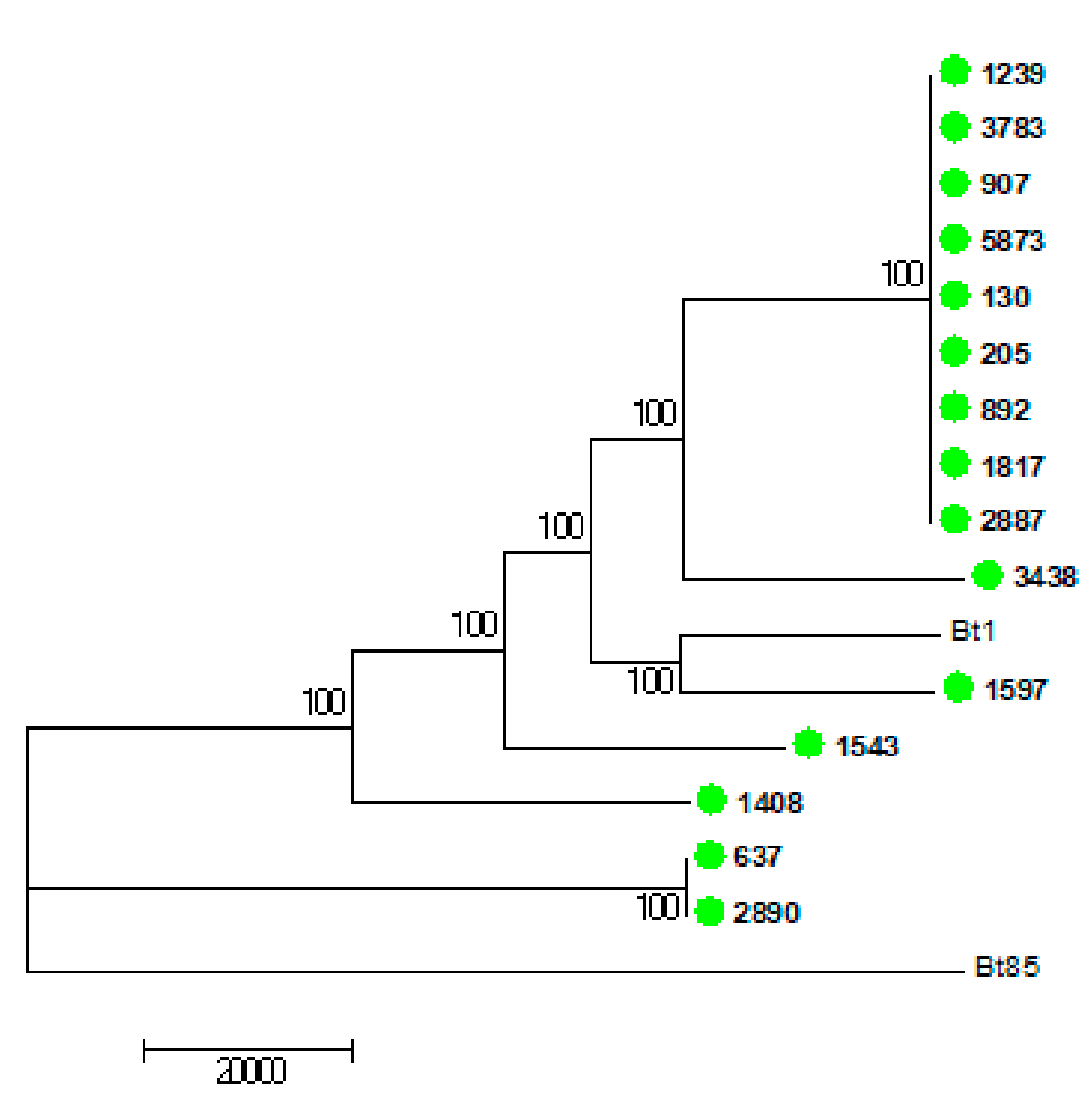
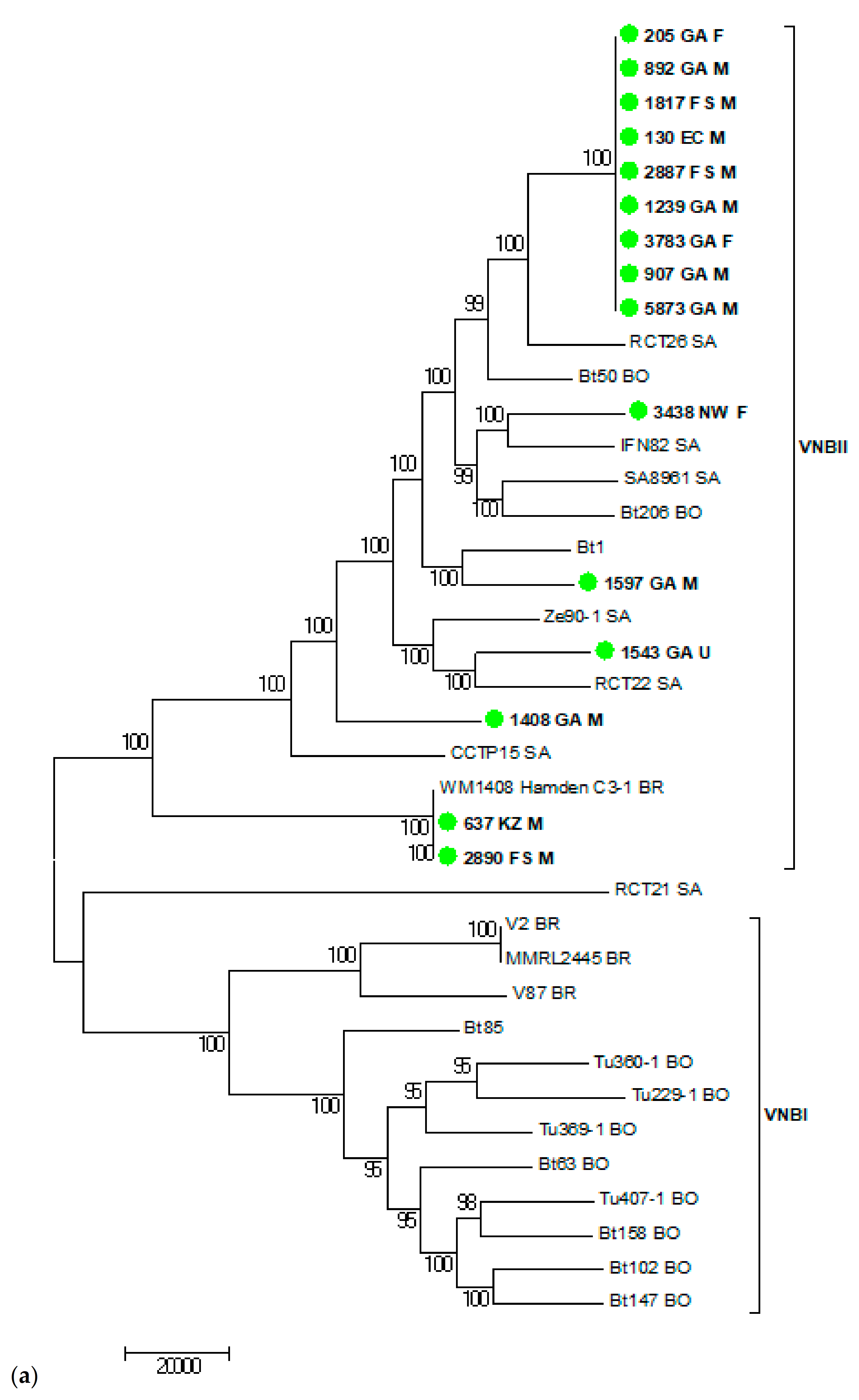
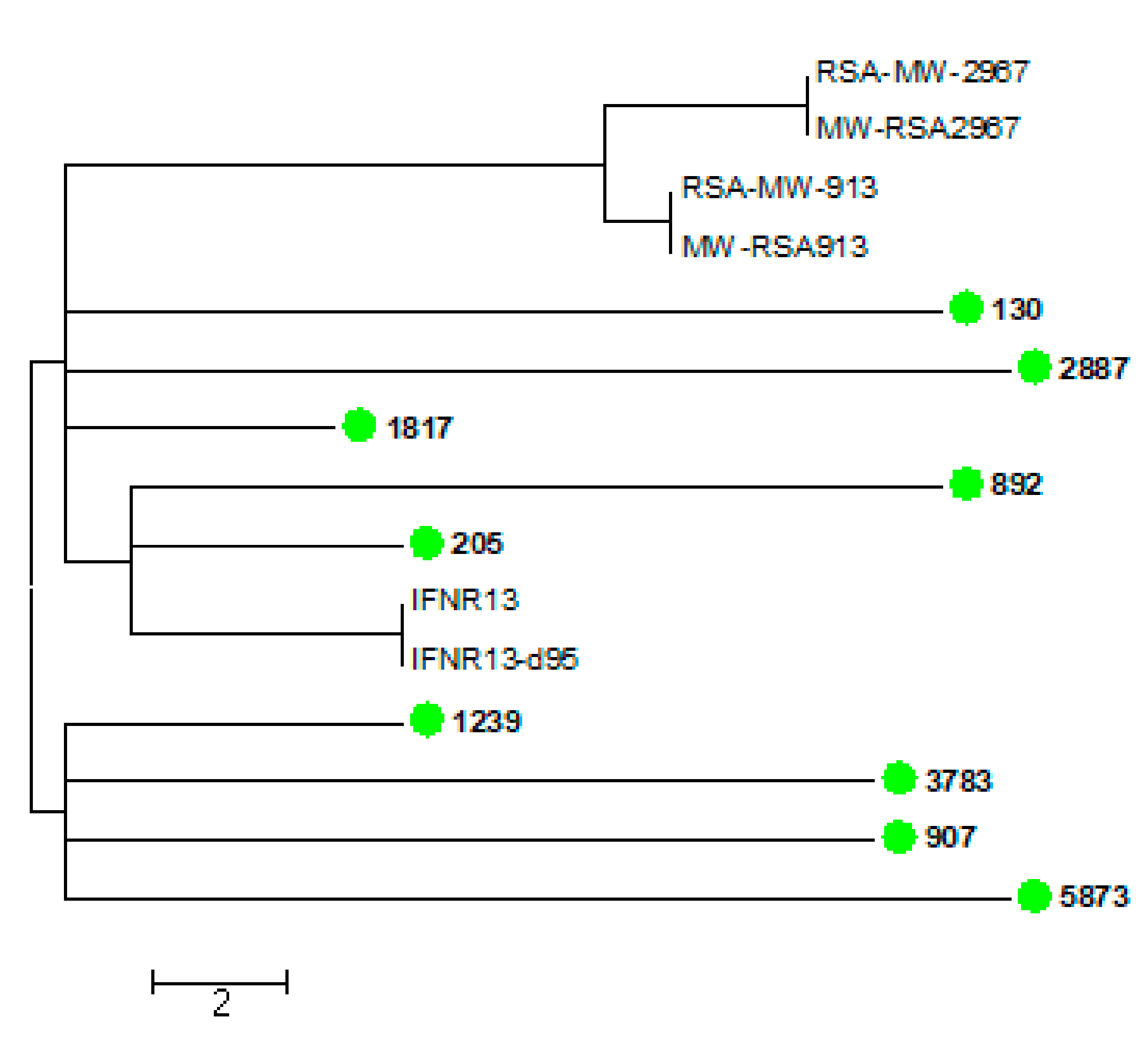
3.6. Virulence and Whole Genome Sequencing (WGS) of 15 C. neoformans Isolates with the VNB Molecular Type
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Britz, E.; Perovic, O.; von Mollendorf, C.; von Gottberg, A.; Iyaloo, S.; Quan, V.; Chetty, V.; Sriruttan, C.; Ismail, N.A.; Nanoo, A.; et al. The Epidemiology of Meningitis among Adults in a South African Province with a High HIV Prevalence, 2009-2012. PLoS ONE 2016, 11, e0163036. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G. Cryptococcal infections in non-HIV-infected patients. Trans. Am. Clin. Climatol. Assoc. 2013, 124, 61–79. [Google Scholar] [PubMed]
- Litvintseva, A.P.; Kestenbaum, L.; Vilgalys, R.; Mitchell, T.G. Comparative analysis of environmental and clinical populations of Cryptococcus neoformans. J. Clin. Microbiol. 2005, 43, 556–564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Govender, N.P.; Meintjes, G.; Mangena, P.; Nel, J.; Potgieter, S.; Reddy, D.; Rabie, H.; Wilson, D.; Black, J.; Boulware, D. Southern African HIV Clinicians Society guideline for the prevention, diagnosis and management of cryptococcal disease among HIV-infected persons: 2019 update. S. Afr. J. HIV Med. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Van Wyk, M.; Govender, N.P.; Mitchell, T.G.; Litvintseva, A.P. Multilocus sequence typing of serially collected isolates of Cryptococcus from HIV-infected patients in South Africa. J. Clin. Microbiol. 2014, 52, 1921. [Google Scholar] [CrossRef]
- Meyer, W.; Aanensen, D.M.; Boekhout, T.; Cogliati, M.; Diaz, M.R.; Esposto, M.C.; Fisher, M.; Gilgado, F.; Hagen, F.; Kaocharoen, S.; et al. Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med. Mycol. 2009, 47, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Bennett, J.E.; Wickes, B.L.; Meyer, W.; Cuomo, C.A.; Wollenburg, K.R.; Bicanic, T.A.; Castaneda, E.; Chang, Y.C.; Chen, J.; et al. The Case for Adopting the “Species Complex” Nomenclature for the Etiologic Agents of Cryptococcosis. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Cogliati, M. Global Molecular Epidemiology of Cryptococcus neoformans and Cryptococcus gattii: An Atlas of the Molecular Types. Sci. Cairo 2013, 2013, 675213. [Google Scholar] [CrossRef]
- Andrade-Silva, L.E.; Ferreira-Paim, K.; Ferreira, T.B.; Vilas-Boas, A.; Mora, D.J.; Manzato, V.M.; Fonseca, F.M.; Buosi, K.; Andrade-Silva, J.; Prudente, B.d.S.; et al. Genotypic analysis of clinical and environmental Cryptococcus neoformans isolates from Brazil reveals the presence of VNB isolates and a correlation with biological factors. PLoS ONE 2018, 13, e0193237. [Google Scholar] [CrossRef]
- Ponzio, V.; Chen, Y.; Rodrigues, A.M.; Tenor, J.L.; Toffaletti, D.L.; Medina-Pestana, J.O.; Colombo, A.L.; Perfect, J.R. Genotypic diversity and clinical outcome of cryptococcosis in renal transplant recipients in Brazil. Emerg. Microbes Infect. 2019, 8, 119–129. [Google Scholar] [CrossRef]
- Litvintseva, A.P.; Carbone, I.; Rossouw, J.; Thakur, R.; Govender, N.P.; Mitchell, T.G. Evidence that the Human Pathogenic Fungus Cryptococcus neoformans var. grubii May Have Evolved in Africa. PLoS ONE 2011, 6, e19688. [Google Scholar] [CrossRef]
- Chen, Y.; Litvintseva, A.P.; Frazzitta, A.E.; Haverkamp, M.R.; Wang, L.; Fang, C.; Muthoga, C.; Mitchell, T.G.; Perfect, J.R. Comparative analyses of clinical and environmental populations of Cryptococcus neoformans in Botswana. Mol. Ecol. 2015, 24, 3559–3571. [Google Scholar] [CrossRef]
- Vanhove, M.; Beale, M.A.; Rhodes, J.; Chanda, D.; Lakhi, S.; Kwenda, G.; Molloy, S.; Karunaharan, N.; Stone, N.; Harrison, T.S.; et al. Genomic epidemiology of Cryptococcus yeasts identifies adaptation to environmental niches underpinning infection across an African HIV/AIDS cohort. Mol. Ecol. 2017, 26, 1991–2005. [Google Scholar] [CrossRef]
- Rhodes, J.; Desjardins, C.A.; Sykes, S.M.; Beale, M.A.; Vanhove, M.; Sakthikumar, S.; Chen, Y.; Gujja, S.; Saif, S.; Chowdhary, A.; et al. Tracing Genetic Exchange and Biogeography of Cryptococcus neoformans var. grubii at the Global Population Level. Genetics 2017, 207, 327–346. [Google Scholar] [CrossRef]
- Desjardins, C.A.; Giamberardino, C.; Sykes, S.M.; Yu, C.H.; Tenor, J.L.; Chen, Y.; Yang, T.; Jones, A.M.; Sun, S.; Haverkamp, M.R.; et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 2017, 27, 1207–1219. [Google Scholar] [CrossRef]
- Beale, M.A.; Sabiiti, W.; Robertson, E.J.; Fuentes-Cabrejo, K.M.; O’Hanlon, S.J.; Jarvis, J.N.; Loyse, A.; Meintjes, G.; Harrison, T.S.; May, R.C.; et al. Genotypic Diversity Is Associated with Clinical Outcome and Phenotype in Cryptococcal Meningitis across Southern Africa. PLoS Negl. Trop. Dis. 2015, 9, e0003847. [Google Scholar] [CrossRef]
- Meiring, S.T.; Quan, V.C.; Cohen, C.; Dawood, H.; Karstaedt, A.S.; McCarthy, K.M.; Whitelaw, A.C.; Govender, N.P. A comparison of cases of paediatric-onset and adult-onset cryptococcosis detected through population-based surveillance, 2005-2007. AIDS Lond. Engl. 2012, 26, 2307–2314. [Google Scholar] [CrossRef]
- Firacative, C.; Roe, C.C.; Malik, R.; Ferreira-Paim, K.; Escandon, P.; Sykes, J.E.; Castanon-Olivares, L.R.; Contreras-Peres, C.; Samayoa, B.; Sorrell, T.C.; et al. MLST and Whole-Genome-Based Population Analysis of Cryptococcus gattii VGIII Links Clinical, Veterinary and Environmental Strains, and Reveals Divergent Serotype Specific Sub-populations and Distant Ancestors. PLoS Negl. Trop. Dis. 2016, 10, e0004861. [Google Scholar] [CrossRef]
- Campbell, L.T.; Currie, B.J.; Krockenberger, M.; Malik, R.; Meyer, W.; Heitman, J.; Carter, D. Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryot. Cell 2005, 4, 1403–1409. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard—Third Edition M27-A3; CLSI: Wayne, PA, USA, 2008. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antifungal Susceptibility Testing of Yeasts—First Edition M60; CLSI: Wayne, PA, USA, 2017. [Google Scholar]
- Naicker, S.D.; Mpembe, R.S.; Maphanga, T.G.; Zulu, T.G.; Desanto, D.; Wadula, J.; Mvelase, N.; Maluleka, C.; Reddy, K.; Dawood, H.; et al. Decreasing fluconazole susceptibility of clinical South African Cryptococcus neoformans isolates over a decade. PLoS Negl. Trop. Dis. 2020, 14, e0008137. [Google Scholar] [CrossRef]
- Espinel-Ingroff, A.; Aller, A.I.; Canton, E.; Castanon-Olivares, L.R.; Chowdhary, A.; Cordoba, S.; Cuenca-Estrella, M.; Fothergill, A.; Fuller, J.; Govender, N.; et al. Cryptococcus neoformans-Cryptococcus gattii species complex: An international study of wild-type susceptibility endpoint distributions and epidemiological cutoff values for fluconazole, itraconazole, posaconazole, and voriconazole. Antimicrob. Agents Chemother 2012, 56, 5898–5906. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Moreno, R.; El Khoury, J.B.; Idnurm, A.; Heitman, J.; Calderwood, S.B.; Ausubel, F.M.; Diener, A. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect. Immun. 2005, 73, 3842–3850. [Google Scholar] [CrossRef] [PubMed]
- Firacative, C.; Duan, S.; Meyer, W. Galleria mellonella model identifies highly virulent strains among all major molecular types of Cryptococcus gattii. PLoS ONE 2014, 9, e105076. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, R.A.C.; Steenwyk, J.L.; Rivero-Menendez, O.; Mead, M.E.; Silva, L.P.; Bastos, R.W.; Alastruey-Izquierdo, A.; Goldman, G.H.; Rokas, A. Genomic and Phenotypic Heterogeneity of Clinical Isolates of the Human Pathogens Aspergillus fumigatus, Aspergillus lentulus, and Aspergillus fumigatiaffinis. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Sahl, J.W.; Lemmer, D.; Travis, J.; Schupp, J.M.; Gillece, J.D.; Aziz, M.; Driebe, E.M.; Drees, K.P.; Hicks, N.D.; Williamson, C.H.D.; et al. NASP: An accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2016, 2, e000074. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, D.L.; Moskalenko, O.; Corcoran, J.M.; McDonald, T.; Rolfes, M.A.; Meya, D.B.; Kajumbula, H.; Kambugu, A.; Bohjanen, P.R.; Knight, J.F. Cryptococcal genotype influences immunologic response and human clinical outcome after meningitis. MBio 2012, 3, e00196-12. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Beale, M.A.; Vanhove, M.; Jarvis, J.N.; Kannambath, S.; Simpson, J.A.; Ryan, A.; Meintjes, G.; Harrison, T.S.; Fisher, M.C.; et al. A Population Genomics Approach to Assessing the Genetic Basis of Within-Host Microevolution Underlying Recurrent Cryptococcal Meningitis Infection. G3 Bethesda Md. 2017, 7, 1165–1176. [Google Scholar] [CrossRef]
- Chen, Y.; Farrer, R.A.; Giamberardino, C.; Sakthikumar, S.; Jones, A.; Yang, T.; Tenor, J.L.; Wagih, O.; Van Wyk, M.; Govender, N.P.; et al. Microevolution of Serial Clinical Isolates of Cryptococcus neoformans var. grubii and C. gattii. MBio 2017, 8. [Google Scholar] [CrossRef]
- Ngouana, T.K.; Drakulovski, P.; Krasteva, D.; Kouanfack, C.; Reynes, J.; Delaporte, E.; Boyom, F.F.; Mallié, M.; Bertout, S. Cryptococcus neoformans isolates from Yaoundé human immunodeficiency virus-infected patients exhibited intra-individual genetic diversity and variation in antifungal susceptibility profiles between isolates from the same patient. J. Med Microbiol. 2016, 65, 579–589. [Google Scholar] [CrossRef][Green Version]
- Kangogo, M.; Bader, O.; Boga, H.; Wanyoike, W.; Folba, C.; Worasilchai, N.; Weig, M.; Groß, U.; Bii, C.C. Molecular types of Cryptococcus gattii/Cryptococcus neoformans species complex from clinical and environmental sources in Nairobi, Kenya. Mycoses 2015, 58, 665–670. [Google Scholar] [CrossRef]
- Kassi, F.K.; Drakulovski, P.; Bellet, V.; Krasteva, D.; Gatchitch, F.; Doumbia, A.; Kouakou, G.A.; Delaporte, E.; Reynes, J.; Mallié, M.; et al. Molecular epidemiology reveals genetic diversity among 363 isolates of the Cryptococcus neoformans and Cryptococcus gattii species complex in 61 Ivorian HIV-positive patients. Mycoses 2016, 59, 811–817. [Google Scholar] [CrossRef]
- Nyazika, T.K.; Herkert, P.F.; Hagen, F.; Mateveke, K.; Robertson, V.J.; Meis, J.F. In vitro antifungal susceptibility profiles of Cryptococcus species isolated from HIV-associated cryptococcal meningitis patients in Zimbabwe. Diagn. Microbiol. Infect. Dis. 2016, 86, 289–292. [Google Scholar] [CrossRef]
- Miglia, K.J.; Govender, N.P.; Rossouw, J.; Meiring, S.; Mitchell, T.G. Analyses of pediatric isolates of Cryptococcus neoformans from South Africa. J. Clin. Microbiol. 2011, 49, 307. [Google Scholar] [CrossRef]
- Ferreira-Paim, K.; Andrade-Silva, L.; Fonseca, F.M.; Ferreira, T.B.; Mora, D.J.; Andrade-Silva, J.; Khan, A.; Dao, A.; Reis, E.C.; Almeida, M.T. MLST-Based Population Genetic Analysis in a Global Context Reveals Clonality amongst Cryptococcus neoformans var. grubii VNI Isolates from HIV Patients in Southeastern Brazil. PLoS Negl. Trop. Dis. 2017, 11, e0005223. [Google Scholar] [CrossRef]
- Lee, G.A.; Arthur, I.; Merritt, A.; Leung, M. Molecular types of Cryptococcus neoformans and Cryptococcus gattii in Western Australia and correlation with antifungal susceptibility. Med. Mycol. 2019, 57, 1004–1010. [Google Scholar] [CrossRef]
- Cogliati, M.; D’Amicis, R.; Zani, A.; Montagna, M.T.; Caggiano, G.; De Giglio, O.; Balbino, S.; De Donno, A.; Serio, F.; Susever, S.; et al. Environmental distribution of Cryptococcus neoformans and C. gattii around the Mediterranean basin. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef]
- Khayhan, K.; Hagen, F.; Pan, W.; Simwami, S.; Fisher, M.C.; Wahyuningsih, R.; Chakrabarti, A.; Chowdhary, A.; Ikeda, R.; Taj-Aldeen, S.J. Geographically structured populations of Cryptococcus neoformans variety grubii in Asia correlate with HIV status and show a clonal population structure. PLoS ONE 2013, 8, e72222. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Type | Total | MIC Value | MIC50 | MIC90 | Geometric Mean | Range | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | 64 | ||||||
| VNI | 60 | 0 | 2 | 10 | 20 | 19 | 3 | 5 | 0 | 1 | 0 | 1 | 6 | 1.43 | 0.25–32 |
| VNB | 15 | 0 | 1 | 2 | 6 | 3 | 3 | 0 | 0 | 0 | 0 | 1 | 4 | 1.26 | 0.25–4 |
| VNII | 25 | 0 | 0 | 5 | 15 | 4 | 1 | 0 | 0 | 0 | 0 | 1 | 2 | 1.03 | 0.5–4 |
| VNIV | 5 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 2 | 2 |
| Total | 105 | 0 | 3 | 17 | 41 | 31 | 7 | 5 | 0 | 1 | 0 | 1 | 4 | 1.32 | 0.25–32 |
| Strain | Median Survival Time | p Value |
|---|---|---|
| H99 * | 2 | NA |
| 5873 | 2 | 0.54 |
| 205 | 3 | 0.02 |
| 2887 | 3 | 0.01 |
| 3438 | 3 | 0.24 |
| 1597 | 3 | 0.01 |
| 907 | 3 | 0.02 |
| 1239 | 3 | 0.04 |
| 3783 | 3 | 0.01 |
| 130 | 3 | 0.01 |
| 1543 | 3 | 0.01 |
| 637 | 4 | 0.02 |
| 1817 | 4 | 0.002 |
| 892 | 4 | 0.003 |
| 2890 | 4 | 0.001 |
| 1408 | 5 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naicker, S.D.; Magobo, R.E.; Maphanga, T.G.; Firacative, C.; van Schalkwyk, E.; Monroy-Nieto, J.; Bowers, J.; Engelthaler, D.M.; Shuping, L.; Meyer, W.; et al. Genotype, Antifungal Susceptibility, and Virulence of Clinical South African Cryptococcus neoformans Strains from National Surveillance, 2005–2009. J. Fungi 2021, 7, 338. https://doi.org/10.3390/jof7050338
Naicker SD, Magobo RE, Maphanga TG, Firacative C, van Schalkwyk E, Monroy-Nieto J, Bowers J, Engelthaler DM, Shuping L, Meyer W, et al. Genotype, Antifungal Susceptibility, and Virulence of Clinical South African Cryptococcus neoformans Strains from National Surveillance, 2005–2009. Journal of Fungi. 2021; 7(5):338. https://doi.org/10.3390/jof7050338
Chicago/Turabian StyleNaicker, Serisha D., Rindidzani E. Magobo, Tsidiso G. Maphanga, Carolina Firacative, Erika van Schalkwyk, Juan Monroy-Nieto, Jolene Bowers, David M. Engelthaler, Liliwe Shuping, Wieland Meyer, and et al. 2021. "Genotype, Antifungal Susceptibility, and Virulence of Clinical South African Cryptococcus neoformans Strains from National Surveillance, 2005–2009" Journal of Fungi 7, no. 5: 338. https://doi.org/10.3390/jof7050338
APA StyleNaicker, S. D., Magobo, R. E., Maphanga, T. G., Firacative, C., van Schalkwyk, E., Monroy-Nieto, J., Bowers, J., Engelthaler, D. M., Shuping, L., Meyer, W., & Govender, N. P. (2021). Genotype, Antifungal Susceptibility, and Virulence of Clinical South African Cryptococcus neoformans Strains from National Surveillance, 2005–2009. Journal of Fungi, 7(5), 338. https://doi.org/10.3390/jof7050338