
Improvement of Torulaspora delbrueckii Genome Annotation: Towards the Exploitation of Genomic Features of a Biotechnologically Relevant Yeast

 , , , ,
, , , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Annotation
2.2. Homology Analysis
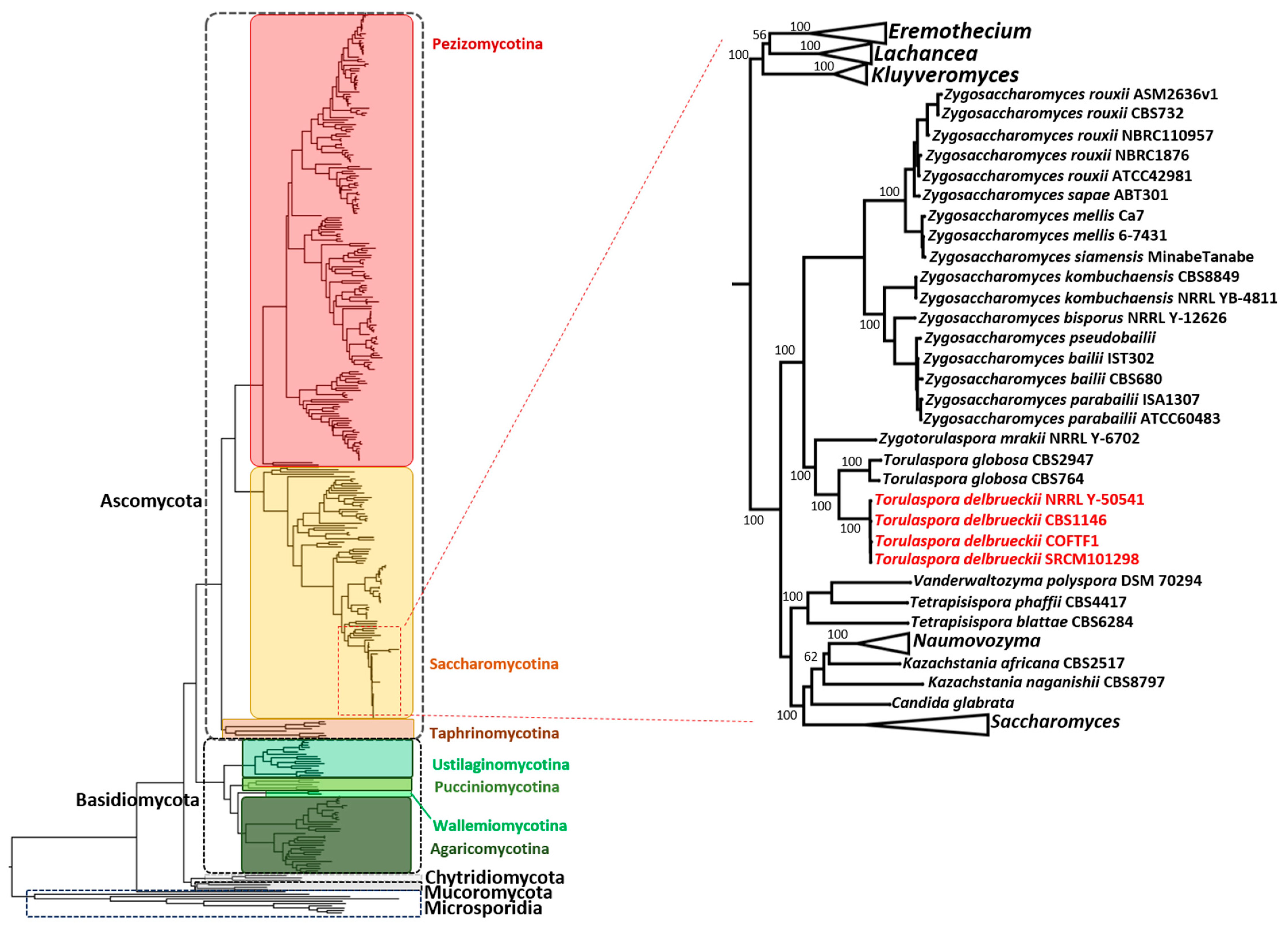
2.3. Phylogenetic Analysis
3. Results and Discussion
3.1. Torulaspora Delbrueckii Genome Annotation
3.2. Functional Annotation
3.3. Homology Analysis
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Domizio, P.; Romani, C.; Lencioni, L.; Comitini, F.; Gobbi, M.; Mannazzu, I.; Ciani, M. Outlining a Future for Non-Saccharomyces Yeasts: Selection of Putative Spoilage Wine Strains to Be Used in Association with Saccharomyces cerevisiae for Grape Juice Fermentation. Int. J. Food Microbiol. 2011, 147, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Tataridis, P.; Kanelis, A.; Logotetis, S.; Nerancis, E. Use of Non-Saccharomyces Torulaspora delbrueckii Yeast Strains in Winemaking and Brewing. Zbornik Matice srpske za prirodne nauke 2013, 415–426. [Google Scholar] [CrossRef]
- Kuanyshev, N.; Adamo, G.M.; Porro, D.; Branduardi, P. The Spoilage Yeast Zygosaccharomyces bailii: Foe or Friend? Yeast 2017, 34, 359–370. [Google Scholar] [CrossRef]
- Vilela, A. Use of Nonconventional Yeasts for Modulating Wine Acidity. Fermentation 2019, 5, 27. [Google Scholar] [CrossRef]
- Bely, M.; Stoeckle, P.; Masneuf-Pomarède, I.; Dubourdieu, D. Impact of Mixed Torulaspora delbrueckii-Saccharomyces cerevisiae Culture on High-Sugar Fermentation. Int. J. Food Microbiol. 2008, 122, 312–320. [Google Scholar] [CrossRef]
- Binati, R.L.; Lemos Junior, W.J.F.; Luzzini, G.; Slaghenaufi, D.; Ugliano, M.; Torriani, S. Contribution of Non-Saccharomyces Yeasts to Wine Volatile and Sensory Diversity: A Study on Lachancea thermotolerans, Metschnikowia spp. and Starmerella bacillaris Strains Isolated in Italy. Int. J. Food Microbiol. 2020, 318, 108470. [Google Scholar] [CrossRef]
- Godoy, L.; Acuña-Fontecilla, A.; Catrileo, D. Formation of Aromatic and Flavor Compounds in Wine: A Perspective of Positive and Negative Contributions of non-Saccharomyces Yeasts. Winemak. Stab. Aging Chem. Biochem. 2020. [Google Scholar] [CrossRef]
- Liu, S.; Laaksonen, O.; Marsol-Vall, A.; Zhu, B.; Yang, B. Comparison of Volatile Composition between Alcoholic Bilberry Beverages Fermented with non-Saccharomyces Yeasts and Dynamic Changes in Volatile Compounds during Fermentation. J. Agric. Food Chem. 2020, 68, 3626–3637. [Google Scholar] [CrossRef]
- Morata, A.; Escott, C.; Bañuelos, M.A.; Loira, I.; del Fresno, J.M.; González, C.; Suárez-lepe, J.A. Contribution of non-Saccharomyces Yeasts to Wine Freshness. A Review. Biomolecules 2020, 10, 34. [Google Scholar] [CrossRef]
- Roca-mesa, H.; Sendra, S.; Mas, A.; Beltran, G.; Torija, M.J. Nitrogen Preferences during Alcoholic Fermentation of Different non-Saccharomyces Yeasts of Oenological Interest. Microorganisms 2020, 8, 157. [Google Scholar] [CrossRef]
- Kosel, J.; Cadež, N.; Schuller, D.; Carreto, L.; Franco-Duarte, R.; Raspor, P. The Influence of Dekkera bruxellensis on the Transcriptome of Saccharomyces cerevisiae and on the Aromatic Profile of Synthetic Wine Must. FEMS Yeast Res. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Pacheco, A.; Santos, J.; Chaves, S.; Almeida, J.; Leo, C.; Joo, M. The Emerging Role of the Yeast Torulaspora delbrueckii in Bread and Wine Production: Using Genetic Manipulation to Study Molecular Basis of Physiological Responses. Struct. Funct. Food Eng. 2012. [Google Scholar] [CrossRef]
- Renault, P.; Coulon, J.; de Revel, G.; Barbe, J.C.; Bely, M. Increase of Fruity Aroma during Mixed T. delbrueckii/S. cerevisiae Wine Fermentation Is Linked to Specific Esters Enhancement; Elsevier B.V.: Amsterdam, The Netherlands, 2015; Volume 207, ISBN 3355757586. [Google Scholar]
- Benito, S. The Impact of Torulaspora Delbrueckii Yeast in Winemaking. Appl. Microbiol. Biotechnol. 2018, 102, 3081–3094. [Google Scholar] [CrossRef] [PubMed]
- Ciani, M.; Picciotti, G. The Growth Kinetics and Fermentation Behaviour of Some non-Saccharomyces Yeasts Associated with Wine-Making. Biotechnol. Lett. 1995, 17, 1247–1250. [Google Scholar] [CrossRef]
- Ciani, M.; Beco, L.; Comitini, F. Fermentation Behaviour and Metabolic Interactions of Multistarter Wine Yeast Fermentations. Int. J. Food Microbiol. 2006, 108, 239–245. [Google Scholar] [CrossRef]
- Tondini, F.; Lang, T.; Chen, L.; Herderich, M.; Jiranek, V. Linking Gene Expression and Oenological Traits: Comparison between Torulaspora delbrueckii and Saccharomyces cerevisiae Strains. Int. J. Food Microbiol. 2019, 294, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mbuyane, L.L.; de Kock, M.; Bauer, F.F.; Divol, B. Torulaspora delbrueckii Produces High Levels of C5 and C6 Polyols during Wine Fermentations. FEMS Yeast Res. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mauricio, J.C.; Millán, C.; Ortega, J.M. Influence of Oxygen on the Biosynthesis of Cellular Fatty Acids, Sterols and Phospholipids during Alcoholic Fermentation by Saccharomyces cerevisiae and Torulaspora delbrueckii. World J. Microbiol. Biotechnol. 1998, 14, 405–410. [Google Scholar] [CrossRef]
- González-Royo, E.; Pascual, O.; Kontoudakis, N.; Esteruelas, M.; Esteve-Zarzoso, B.; Mas, A.; Canals, J.M.; Zamora, F. Oenological Consequences of Sequential Inoculation with non-Saccharomyces Yeasts (Torulaspora delbrueckii or Metschnikowia pulcherrima) and Saccharomyces cerevisiae in Base Wine for Sparkling Wine Production. Eur. Food Res. Technol. 2015, 240, 999–1012. [Google Scholar] [CrossRef]
- Benito, Á.; Calderón, F.; Benito, S. The Influence of non-Saccharomyces Species on Wine Fermentation Quality Parameters. Fermentation 2019, 5, 54. [Google Scholar] [CrossRef]
- Almeida, M.J.; Pais, C. Leavening Ability and Freeze Tolerance of Yeasts Isolated from Traditional Corn and Rye Bread Doughs. Appl. Environ. Microbiol. 1996, 62, 4401–4404. [Google Scholar] [CrossRef]
- Hernandez-Lopez, M.J.; Prieto, J.A.; Randez-Gil, F. Osmotolerance and Leavening Ability in Sweet and Frozen Sweet Dough. Comparative Analysis between Torulaspora delbrueckii and Saccharomyces cerevisiae Baker’s Yeast Strains. Antonie van Leeuwenhoek. Int. J. Gen. Mol. Microbiol. 2003, 84, 125–134. [Google Scholar] [CrossRef]
- Alves-Araújo, C.; Almeida, M.J.; Sousa, M.J.; Leão, C. Freeze Tolerance of the Yeast Torulaspora delbrueckii: Cellular and Biochemical Basis. FEMS Microbiol. Lett. 2004, 240, 7–14. [Google Scholar] [CrossRef]
- Santos, J.; Sousa, M.J.; Cardoso, H.; Inácio, J.; Silva, S.; Spencer-Martins, I.; Leão, C. Ethanol Tolerance of Sugar Transport, and the Rectification of Stuck Wine Fermentations. Microbiology 2008, 154, 422–430. [Google Scholar] [CrossRef]
- Rachidi, N.; Martinez, M.J.; Barre, P.; Blondin, B. Saccharomyces cerevisiae PAU Genes Are Induced by Anaerobiosis. Mol. Microbiol. 2000, 35, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Steensels, J.; Verstrepen, K.J. Taming Wild Yeast: Potential of Conventional and Nonconventional Yeasts in Industrial Fermentations. Annu. Rev. Microbiol. 2014, 68, 61–80. [Google Scholar] [CrossRef]
- Canonico, L.; Agarbati, A.; Comitini, F.; Ciani, M. Torulaspora delbrueckii in the Brewing Process: A New Approach to Enhance Bioflavour and to Reduce Ethanol Content. Food Microbiol. 2016, 56, 45–51. [Google Scholar] [CrossRef]
- Visintin, S.; Ramos, L.; Batista, N.; Dolci, P.; Schwan, F.; Cocolin, L. Impact of Saccharomyces cerevisiae and Torulaspora delbrueckii Starter Cultures on Cocoa Beans Fermentation. Int. J. Food Microbiol. 2017, 257, 31–40. [Google Scholar] [CrossRef]
- Clemente-Jimenez, J.M.; Mingorance-Cazorla, L.; Martínez-Rodríguez, S.; las Heras-Vázquez, F.J.; Rodríguez-Vico, F. Influence of Sequential Yeast Mixtures on Wine Fermentation. Int. J. Food Microbiol. 2005, 98, 301–308. [Google Scholar] [CrossRef]
- Belda, I.; Ruiz, J.; Beisert, B.; Navascués, E.; Marquina, D.; Calderón, F.; Rauhut, D.; Benito, S.; Santos, A. Influence of Torulaspora delbrueckii in Varietal Thiol (3-SH and 4-MSP) Release in Wine Sequential Fermentations. Int. J. Food Microbiol. 2017, 257, 183–191. [Google Scholar] [CrossRef]
- Padilla, B.; Zulian, L.; Ferreres, À.; Pastor, R.; Esteve-Zarzoso, B.; Beltran, G.; Mas, A. Sequential Inoculation of Native non-Saccharomyces and Saccharomyces cerevisiae Strains for Wine Making. Front. Microbiol. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Sanoppa, K.; Huang, T.C.; Wu, M.C. Effects of Saccharomyces cerevisiae in Association with Torulaspora delbrueckii on the Aroma and Amino Acids in Longan Wines. Food Sci. Nutr. 2019, 7, 2817–2826. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The Genomics Resource of Budding Yeast. Nucleic Acids Res. 2012, 40, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L.; Armisen, D.; Proux-Weŕa, E.; ÓhEígeartaigh, S.S.; Byrne, K.P.; Wolfe, K.H. Evolutionary Erosion of Yeast Sex Chromosomes by Mating-Type Switching Accidents. Proc. Natl. Acad. Sci. USA 2011, 108, 20024–20029. [Google Scholar] [CrossRef]
- Tondini, F.; Jiranek, V.; Grbin, P.R.; Onetto, C.A. Genome Sequence of Australian Indigenous Wine Yeast Torulaspora delbrueckii COFT1 Using Nanopore Sequencing. Genome Announc. 2018, 6, 1–2. [Google Scholar] [CrossRef]
- Gomez-Angulo, J.; Vega-Alvarado, L.; Escalante-García, Z.; Grande, R.; Gschaedler-Mathis, A.; Amaya-Delgado, L.; Arrizon, J.; Sanchez-Flores, A. Genome Sequence of Torulaspora delbrueckii NRRL Y-50541, Isolated from Mezcal Fermentation. Genome Announc. 2015, 3, 3–4. [Google Scholar] [CrossRef]
- Coughlan, A.Y.; Lombardi, L.; Braun-Galleani, S.; Martos, A.A.R.; Galeote, V.; Bigey, F.; Dequin, S.; Byrne, K.P.; Wolfe, K.H. The Yeast Mating-Type Switching Endonuclease HO Is a Domesticated Member of an Unorthodox Homing Genetic Element Family. eLife 2020, 9, 1–24. [Google Scholar] [CrossRef]
- Proux-Wéra, E.; Armisén, D.; Byrne, K.P.; Wolfe, K.H. A Pipeline for Automated Annotation of Yeast Genome Sequences by a Conserved-Synteny Approach. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Methods in Molecular Biology; SBL Press: Williston, VT, USA, 2019; Volume 1962, pp. 227–245. ISBN 9781493991730. [Google Scholar]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by EggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, Information, Knowledge and Principle: Back to Metabolism in KEGG. Nucleic Acids Res. 2014, 42, 199–205. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial Genome Coverage and Improved Protein Family Annotation in the COG Database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for Making Accurate Alignments of Many Protein Sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Gabaldón, T.; Koonin, E.V. Functional and Evolutionary Implications of Gene Orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Diderich, J.A.; Schepper, M.; van Hoek, P.; Luttik, M.A.H.; van Dijken, J.P.; Pronk, J.T.; Klaassen, P.; Boelens, H.F.M.; de Mattos, M.J.T.; van Dam, K.; et al. Glucose Uptake Kinetics and Transcription of HXT Genes in Chemostat Cultures of Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 15350–15359. [Google Scholar] [CrossRef]
- Ye, L.; Kruckeberg, A.L.; Berden, J.A.; van Dam, K. Growth and Glucose Repression Are Controlled by Glucose Transport in Saccharomyces cerevisiae Cells Containing Only One Glucose Transporter. J. Bacteriol. 1999, 181, 4673–4675. [Google Scholar] [CrossRef]
- Franco-Duarte, R.; Bigey, F.; Carreto, L.; Mendes, I.; Dequin, S.; Santos, M.A.S.; Pais, C.; Schuller, D. Intrastrain Genomic and Phenotypic Variability of the Commercial Saccharomyces cerevisiae Strain Zymaflore VL1 Reveals Microevolutionary Adaptation to Vineyard Environments. FEMS Yeast Res. 2015, 15, 1–13. [Google Scholar] [CrossRef][Green Version]
- Alves-Araújo, C.; Hernandez-Lopez, M.J.; Prieto, J.A.; Randez-Gil, F.; Sousa, M.J. Isolation and Characterization of the LGT1 Gene Encoding a Low-Affinity Glucose Transporter from Torulaspora delbrueckii. Yeast 2005, 22, 165–175. [Google Scholar] [CrossRef]
- Pacheco, A.; Donzella, L.; Hernandez-Lopez, M.J.; Almeida, M.J.; Prieto, J.A.; Randez-Gil, F.; Morrissey, J.P.; Sousa, M.J. Hexose Transport in Torulaspora delbrueckii: Identification of Igt1, a New Dual-Affinity Transporter. FEMS Yeast Res. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Petruzzi, L.; Capozzi, V.; Berbegal, C.; Corbo, M.R.; Bevilacqua, A.; Spano, G.; Sinigaglia, M. Microbial Resources and Enological Significance: Opportunities and Benefits. Front. Microbiol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nadai, C.; Treu, L.; Campanaro, S.; Giacomini, A.; Corich, V. Different Mechanisms of Resistance Modulate Sulfite Tolerance in Wine Yeasts. Appl. Microbiol. Biotechnol. 2016, 100, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Franco-Duarte, R.; Umek, L.; Mendes, I.; Castro, C.C.; Fonseca, N.; Martins, R.; Silva-Ferreira, A.C.; Sampaio, P.; Pais, C.; Schuller, D. New Integrative Computational Approaches Unveil the Saccharomyces cerevisiae Pheno-Metabolomic Fermentative Profile and Allow Strain Selection for Winemaking. Food Chem. 2016, 211, 509–520. [Google Scholar] [CrossRef]
- Franco-Duarte, R.; Mendes, I.; Umek, L.; Drumonde-Neves, J.; Zupan, B.; Schuller, D. Computational Models Reveal Genotype-Phenotype Associations in Saccharomyces cerevisiae. Yeast 2014, 31, 265–277. [Google Scholar] [CrossRef]
- Wong, S.; Wolfe, K.H. Duplication of Genes and Genomes in Yeasts. Top. Curr. Genet. 2006, 15, 79–99. [Google Scholar] [CrossRef]
- Wolfe, K.H. Origin of the Yeast Whole-Genome Duplication. PLoS Biol. 2015, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- James, S.A.; Collins, M.D.; Roberts, I.N. Use of an RRNA Internal Transcribed Spacer Region to Distinguish Phylogenetically Closely Related Species of the Genera Zygosaccharomyces and Torulaspora. Int. J. Syst. Bacteriol. 1996, 46, 189–194. [Google Scholar] [CrossRef]
- Aller-Arranz, E.; Randez-Gil, F.; Barrio, E.; Prieto, J.A. A DNA Region of Torulaspora delbrueckii Containing the HIS3 Gene: Sequence, Gene Order and Evolution. Yeast 2003, 20, 1359–1368. [Google Scholar] [CrossRef]
- Escribano, R.; González-Arenzana, L.; Portu, J.; Garijo, P.; López-Alfaro, I.; López, R.; Santamaría, P.; Gutiérrez, A.R. Wine Aromatic Compound Production and Fermentative Behaviour within Different non-Saccharomyces Species and Clones. J. Appl. Microbiol. 2018, 124, 1521–1531. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Phylogenetic Relationships among Yeasts of the “Saccharomyces Complex” Determined from Multigene Sequence Analyses. FEMS Yeast Res. 2003, 3, 417–432. [Google Scholar] [CrossRef]
- Kurtzman, C.P. Phylogenetic Circumscription of Saccharomyces, Kluyveromyces and Other Members of the Saccharomycetaceae, and the Proposal of the New Genera Lachancea, Nakaseomyces, Naumovia, Vanderwaltozyma and Zygotorulaspora. FEMS Yeast Res. 2003, 4, 233–245. [Google Scholar] [CrossRef]
- Domizio, P.; House, J.F.; Joseph, C.M.L.; Bisson, L.F.; Bamforth, C.W. Lachancea Thermotolerans as an Alternative Yeast for the Production of Beer. J. Inst. Brew. 2016, 122, 599–604. [Google Scholar] [CrossRef]
- Benito, Á.; Calderón, F.; Palomero, F.; Benito, S. Quality and Composition of Airén Wines Fermented by Sequential Inoculation of Lachancea thermotolerans and Saccharomyces cerevisiae. Food Technol. Biotechnol. 2016, 54, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Thompson Witrick, K.; Duncan, S.; Hurley, K.; O’Keefe, S. Acid and Volatiles of Commercially-Available Lambic Beers. Beverages 2017, 3, 51. [Google Scholar] [CrossRef]
- Toh, D.W.K.; Chua, J.Y.; Lu, Y.; Liu, S.Q. Evaluation of the Potential of Commercial non-Saccharomyces Yeast Strains of Torulaspora delbrueckii and Lachancea thermotolerans in Beer Fermentation. Int. J. Food Sci. Technol. 2020, 55, 2049–2059. [Google Scholar] [CrossRef]
- Shen, X.X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.B.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545.e20. [Google Scholar] [CrossRef]
- Roetzer, A.; Gabaldón, T.; Schüller, C. From Saccharomyces cerevisiae to Candida glabrata in a Few Easy Steps: Important Adaptations for an Opportunistic Pathogen. FEMS Microbiol. Lett. 2011, 314, 1–9. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Source | Reference | Genome Size | BUSCO Genome Completeness (%) | Coding Sequences | Transposable Elements (TY) | tRNA Genes | Homologies with S. cerevisiae | Unidentified Coding Sequences 1 |
|---|---|---|---|---|---|---|---|---|---|
| CBS1146 | Unknown; type strain | [35] | 9.22 Mb | 98.5 | 4978 | 5 | 191 | 4514 | 464 |
| COFT1 | Wine fermentations | [36] | 9.36 Mb | 98.1 | 5009 | 5 | 223 | 4506 | 503 |
| NRRL Y-50541 | Mezcal-fermentations | [37] | 11.53 Mb | 80.2 | 4361 | 6 | 180 | 3875 | 486 |
| SRCM101298 | Fermented food | - | 9.68 Mb | 98.2 | 5016 | 7 | 274 | 4513 | 503 |
| CBS1146 | COFT1 | NRRL Y-50541 | |
|---|---|---|---|
| CBS1146 | |||
| COFT1 | 99.54 (±0.71) | ||
| NRRL Y-50541 | 97.98 (±2.28) | 97.89 (±2.29) | |
| SCRM101298 | 99.54 (±0.74) | 99.63 (±0.66) | 97.62 (±2.29) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago, C.; Rito, T.; Vieira, D.; Fernandes, T.; Pais, C.; Sousa, M.J.; Soares, P.; Franco-Duarte, R. Improvement of Torulaspora delbrueckii Genome Annotation: Towards the Exploitation of Genomic Features of a Biotechnologically Relevant Yeast. J. Fungi 2021, 7, 287. https://doi.org/10.3390/jof7040287
Santiago C, Rito T, Vieira D, Fernandes T, Pais C, Sousa MJ, Soares P, Franco-Duarte R. Improvement of Torulaspora delbrueckii Genome Annotation: Towards the Exploitation of Genomic Features of a Biotechnologically Relevant Yeast. Journal of Fungi. 2021; 7(4):287. https://doi.org/10.3390/jof7040287
Chicago/Turabian StyleSantiago, Carolina, Teresa Rito, Daniel Vieira, Ticiana Fernandes, Célia Pais, Maria João Sousa, Pedro Soares, and Ricardo Franco-Duarte. 2021. "Improvement of Torulaspora delbrueckii Genome Annotation: Towards the Exploitation of Genomic Features of a Biotechnologically Relevant Yeast" Journal of Fungi 7, no. 4: 287. https://doi.org/10.3390/jof7040287
APA StyleSantiago, C., Rito, T., Vieira, D., Fernandes, T., Pais, C., Sousa, M. J., Soares, P., & Franco-Duarte, R. (2021). Improvement of Torulaspora delbrueckii Genome Annotation: Towards the Exploitation of Genomic Features of a Biotechnologically Relevant Yeast. Journal of Fungi, 7(4), 287. https://doi.org/10.3390/jof7040287