Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Isolation
2.2. DNA Extraction and WGS Library Construction
2.3. High-Through Chromosome Conformation Capture (Hi-C) Library Construction
2.4. Genome Sequencing and Assembly
2.5. Identification of Repetitive Elements and Non-Coding RNA Genes
2.6. Gene Prediction and Function Annotation
2.7. Core Genes, Homolog Genes, SNPs and Phylogeny Reconstruction
3. Results and Discussion
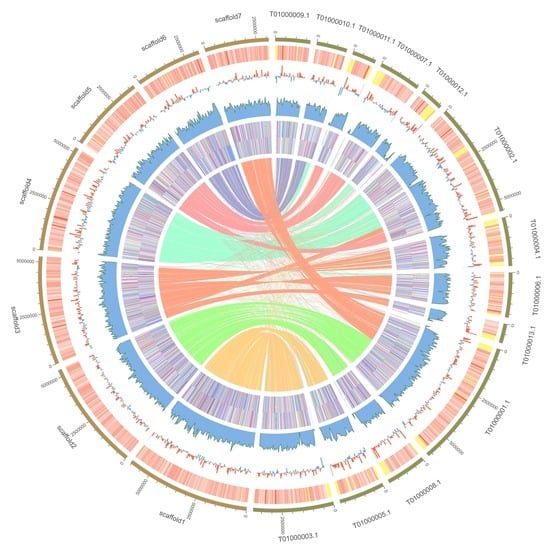
3.1. Genome Assembly
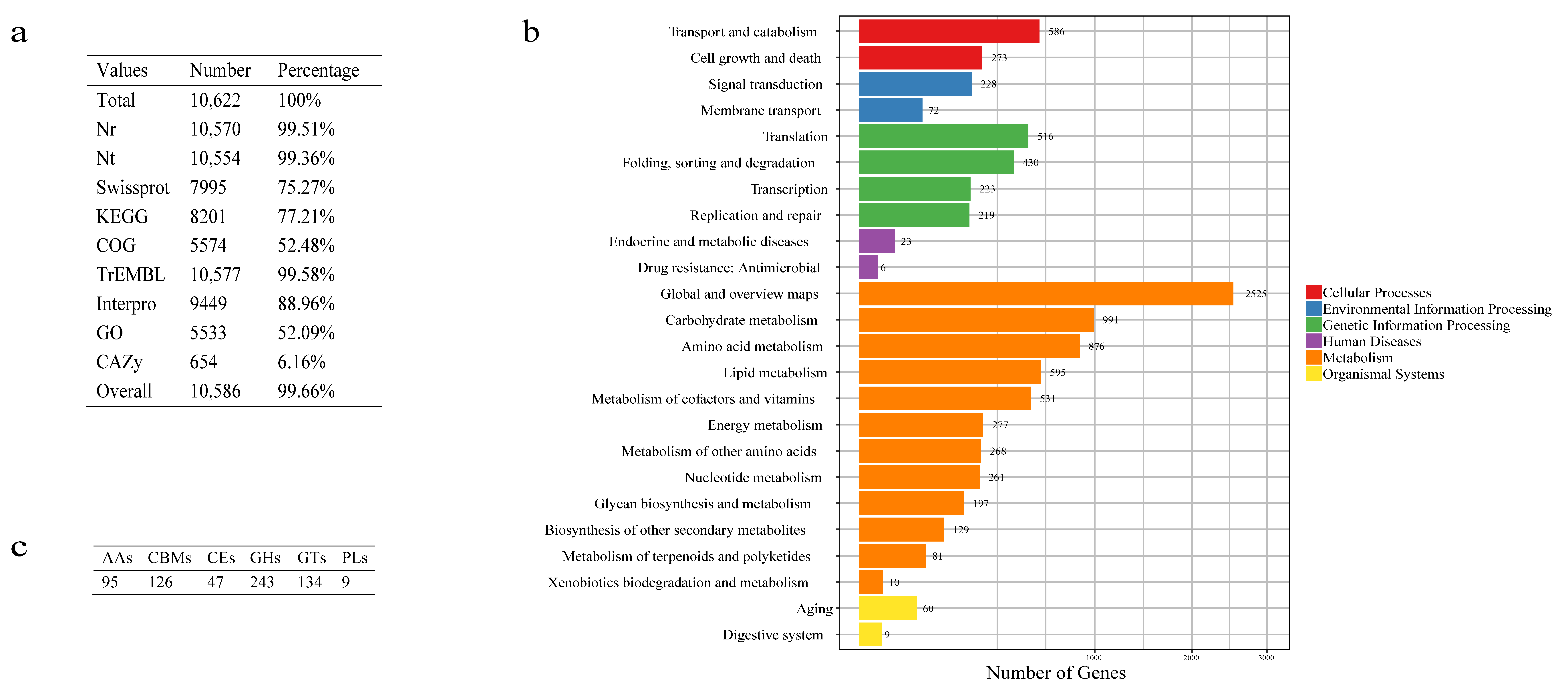
3.2. Genome Annotation
3.3. Functional Categorization
3.4. Genome Comparison of P. variabile and T.islandicus
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, C.S.; Li, X.M.; Gao, S.S.; Lu, Y.H.; Wang, B.G. Cytotoxic anthranilic acid derivatives from deep sea sediment-derived fungus penicillium paneum sd-44. Mar. Drugs 2013, 11, 3068–3076. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.H.; Zhang, P.; Li, X.M.; Wang, B.G. Penicibrocazines a–e, five new sulfide diketopiperazines from the marine-derived endophytic funguspenicillium brocae. Mar. Drugs 2015, 13, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Su, M.; Song, S.J.; Jung, J.H. Marine-derived penicillium species as producers of cytotoxic metabolites. Mar. Drugs 2017, 15, 329. [Google Scholar] [CrossRef] [PubMed]
- Latha, R.; Mitra, S.J.C.S. Mangrove fungi in india. Curr. Sci. 2004, 86, 1586. [Google Scholar]
- Hrudayanath, T.; Bikash, C.B.; Rashmi, R.M. Ecological role and biotechnological potential of mangrove fungi: A review. Mycobiology 2013, 4, 54–71. [Google Scholar]
- Zhang, Z.; He, X.; Zhang, G.; Che, Q.; Zhu, T.; Gu, Q.; Li, D. Inducing secondary metabolite production by combined culture of talaromyces aculeatus and penicillium variabile. J. Nat. Prod. 2017, 80, 3167–3171. [Google Scholar] [CrossRef]
- He, X.; Zhang, Z.; Chen, Y.; Che, Q.; Zhu, T.; Gu, Q.; Li, D. Varitatin a, a highly modified fatty acid amide from penicillium variabile cultured with a DNA methyltransferase inhibitor. J. Nat. Prod. 2015, 78, 2841–2845. [Google Scholar] [CrossRef]
- Zhelifonova, V.P.; Antipova, T.V.; Ozerskaya, S.M.; Ivanushkina, N.E.; Kozlovskii, A.G. The fungus penicillium variabile sopp 1912 isolated from permafrost deposits as a producer of rugulovasines. Microbiology 2006, 75, 644–648. [Google Scholar] [CrossRef]
- Dorner, J.; Cole, R.; Hill, R.; Wicklow, D.; Cox, R.H. Penicillium rubrum and penicillium biforme, new sources of rugulovasines a and b. Appl. Environ. Microbiol. 1980, 40, 685–687. [Google Scholar]
- Meurant, G. Handbook of Toxic Fungal Metabolites; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Gu, X.-L.; Tian, Y.P. Isolation and characterization of urethanase from penicillium variabile and its application to reduce ethyl carbamate contamination in chinese rice wine. Appl. Biochem. Biotechnol. 2013, 170, 718–728. [Google Scholar]
- Petruccioli, M.; Federici, F.; Bucke, C.; Keshavarz, T. Enhancement of glucose oxidase production by penicillium variabile p16. Enzym. Microb. Technol. 1999, 24, 397–401. [Google Scholar] [CrossRef]
- Schafhauser, T.; Wibberg, D.; Ruckert, C.; Winkler, A.; Flor, L.; van Pee, K.H.; Fewer, D.P.; Sivonen, K.; Jahn, L.; Ludwig-Muller, J.; et al. Draft genome sequence of talaromyces islandicus (“penicillium islandicum”) wf-38-12, a neglected mold with significant biotechnological potential. J. Biotechnol. 2015, 211, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Marcet-Houben, M.; Ballester, A.R.; de la Fuente, B.; Harries, E.; Marcos, J.F.; Gonzalez-Candelas, L.; Gabaldon, T. Genome sequence of the necrotrophic fungus penicillium digitatum, the main postharvest pathogen of citrus. BMC Genom. 2012, 13, 646. [Google Scholar] [CrossRef] [PubMed]
- Drmanac, R.; Sparks, A.B.; Callow, M.J.; Halpern, A.L.; Burns, N.L.; Kermani, B.G.; Carnevali, P.; Nazarenko, I.; Nilsen, G.B.; Yeung, G.; et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 2010, 327, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. Soapnuke: A mapreduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. Hic-pro: An optimized and flexible pipeline for hi-c data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. , et al. De novo assembly of the aedes aegypti genome using hi-c yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Harris, R. Improved Pairwise Alignment of Genomic DNA: The Pennsylvania State University. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using repeatmasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. Ltr_finder: An efficient tool for the prediction of full-length ltr retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. Trnascan-se: A program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Cui, X.; Lu, Z.; Wang, S.; Jing-Yan Wang, J.; Gao, X. Cmsearch: Simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016, 32, i332–i340. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-coding rna analysis using the rfam database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef]
- Kent, W.J. Blat--the blast-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. Genewise and genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. Augustus: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. Genemarks: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Mackey, A.J.; Reese, J.T.; Milshina, N.V.; Roos, D.S.; Weinstock, G.M. Creating a honey bee consensus gene set. Genome Biol. 2007, 8, R13. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Reorganizing the protein space at the universal protein resource (uniprot). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar]
- Bairoch, A.; Apweiler, R. The swiss-prot protein sequence database and its supplement trembl in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The cog database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (cazy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. Ncbi reference sequence (refseq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Ponting, C.P.; Schultz, J.; Milpetz, F.; Bork, P. Smart: Identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999, 27, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Lazareva-Ulitsky, B.; Loo, R.; Kejariwal, A.; Vandergriff, J.; Rabkin, S.; Guo, N.; Muruganujan, A.; Doremieux, O.; Campbell, M.J.; et al. The panther database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33, D284–D288. [Google Scholar] [CrossRef] [PubMed]
- Attwood, T.K. The prints database: A resource for identification of protein families. Brief. Bioinform. 2002, 3, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Hulo, N.; Bairoch, A.; Bulliard, V.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Pagni, M.; Sigrist, C.J. The prosite database. Nucleic Acids Res. 2006, 34, D227–D230. [Google Scholar] [CrossRef]
- Bru, C.; Courcelle, E.; Carrere, S.; Beausse, Y.; Dalmar, S.; Kahn, D. The prodom database of protein domain families: More emphasis on 3d. Nucleic Acids Res. 2005, 33, D212–D215. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. Interproscan--an integration platform for the signature-recognition methods in interpro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. Antismash: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016, 17, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Chacko, N.; Lin, X. Non-coding rnas in the development and pathogenesis of eukaryotic microbes. Appl. Microbiol. Biotechnol. 2013, 97, 7989–7997. [Google Scholar] [CrossRef]
- Keller, N.P.; Bennett, J.; Turner, G. Secondary metabolism: Then, now and tomorrow. Fungal Genet. Biol. 2011, 48, 1–3. [Google Scholar] [CrossRef]
- Bills, G.F.; Gloer, J.B. Biologically active secondary metabolites from the fungi. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Ancheeva, E.; Daletos, G.; Proksch, P. Lead compounds from mangrove-associated microorganisms. Mar. Drugs 2018, 16, 319. [Google Scholar] [CrossRef]
- Yilmaz, N.; Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of the genus talaromyces. Stud. Mycol. 2014, 78, 175–341. [Google Scholar] [CrossRef]
- Chen, A.J.; Sun, B.D.; Houbraken, J.; Frisvad, J.C.; Yilmaz, N.; Zhou, Y.G.; Samson, R.A. New talaromyces species from indoor environments in china. Stud. Mycol. 2016, 84, 119–144. [Google Scholar] [CrossRef]
- Dupont, J.; Dennetière, B.; Jacquet, C.; Roquebert, M.-F. Pcr-rflp of its rdna for the rapid identification of penicillium subgenus biverticillium species. Rev. Iberoam. Micol. 2006, 23, 145–150. [Google Scholar] [CrossRef]
- Prat, C.; Ruiz-Rueda, O.; Trias, R.; Antico, E.; Capone, D.; Sefton, M.; Baneras, L. Molecular fingerprinting by pcr-denaturing gradient gel electrophoresis reveals differences in the levels of microbial diversity for musty-earthy tainted corks. Appl. Environ. Microbiol. 2009, 75, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Chen, J.; Jin, T.; Shi, C.; Du, X.; Zhang, H.; Zhang, Y.; Li, H.; Luo, T.; Yan, P.; et al. The report of marine life genomic research. Preprints 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Values | Draft Genome | Chromosome Genome | ||
|---|---|---|---|---|
| Scaffold | Contig | Scaffold | Contig | |
| Num | 3860 | 3860 | 348 | 757 |
| Length (bp) | 34,059,084 | 34,059,084 | 33,318,899 | 33,114,399 |
| N50 (bp) | 93,482 | 93,482 | 5,232,202 | 96,738 |
| N90 (bp) | 24,393 | 24,393 | 2,951,000 | 30,747 |
| GC% | 46.58 | 47 | 48 | 48 |
| Values | Homolog | denovo | Glean | |||
|---|---|---|---|---|---|---|
| P. expansum | P. rubens | T. marneffei | T. stipitatus | Augustus | ||
| Gene_number | 8947 | 9214 | 9510 | 9360 | 12,032 | 10,622 |
| Average_gene_len (bp) | 1456.62 | 1394.67 | 1459.34 | 1478.38 | 1773.88 | 1994.87 |
| Average_cds_len (bp) | 1317.95 | 1294.49 | 1349.26 | 1363.3 | 1581.44 | 1568.29 |
| Average_exon_number | 2.34 | 2.31 | 2.43 | 2.47 | 3.38 | 3.31 |
| Average_exon_len (bp) | 562.45 | 560.76 | 554.42 | 552.59 | 468.3 | 473.94 |
| Average_intron_len (bp) | 103.24 | 76.57 | 76.78 | 78.44 | 80.96 | 184.74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, L.; Li, L.; Liu, X.; Chen, J.; Shi, C.; Guo, W.; Xu, Q.; Fan, G.; Liu, X.; Li, D. Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1. J. Fungi 2020, 6, 7. https://doi.org/10.3390/jof6010007
Peng L, Li L, Liu X, Chen J, Shi C, Guo W, Xu Q, Fan G, Liu X, Li D. Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1. Journal of Fungi. 2020; 6(1):7. https://doi.org/10.3390/jof6010007
Chicago/Turabian StylePeng, Ling, Liangwei Li, Xiaochuan Liu, Jianwei Chen, Chengcheng Shi, Wenjie Guo, Qiwu Xu, Guangyi Fan, Xin Liu, and Dehai Li. 2020. "Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1" Journal of Fungi 6, no. 1: 7. https://doi.org/10.3390/jof6010007
APA StylePeng, L., Li, L., Liu, X., Chen, J., Shi, C., Guo, W., Xu, Q., Fan, G., Liu, X., & Li, D. (2020). Chromosome-Level Comprehensive Genome of Mangrove Sediment-Derived Fungus Penicillium variabile HXQ-H-1. Journal of Fungi, 6(1), 7. https://doi.org/10.3390/jof6010007