Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
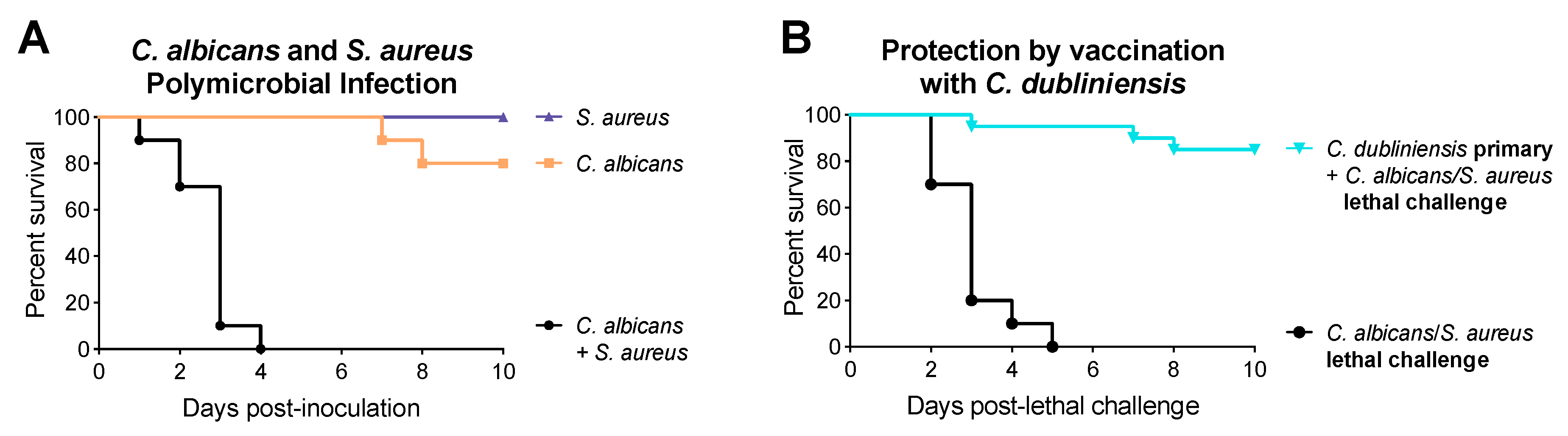
1.1. Synergism between Candida and Bacteria
1.2. C. dubliniensis-Mediated Protection against Polymicrobial Sepsis
2. Myeloid-Derived Suppressor Cells
2.1. MDSC Subsets
2.2. Development of MDSCs
2.3. Mechanisms of MDSC Immunosuppression
2.4. Limitations of Studying MDSCs
3. Role of MDSCs in Sepsis and Infection
4. Recent Advances in MDSC-Mediated Trained Innate Immunity against Polymicrobial IAI
4.1. Properties of C. dubliniensis-Mediated Trained Innate Immune Protection
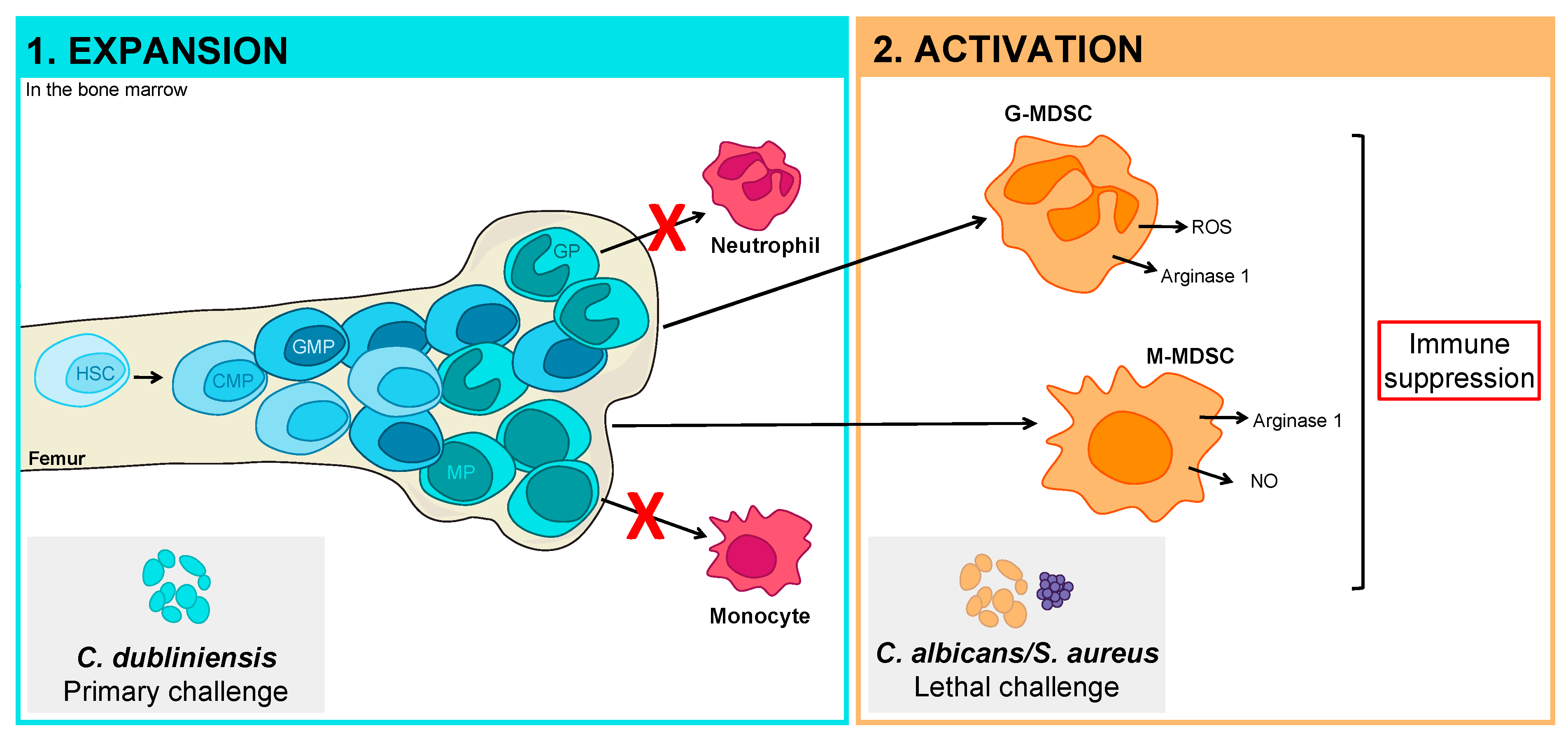
4.2. Pathogen Manipulation of the Hematopoietic Compartment
5. Perspectives
5.1. Development of Pathogen-Specific MDSCs of Limited Function
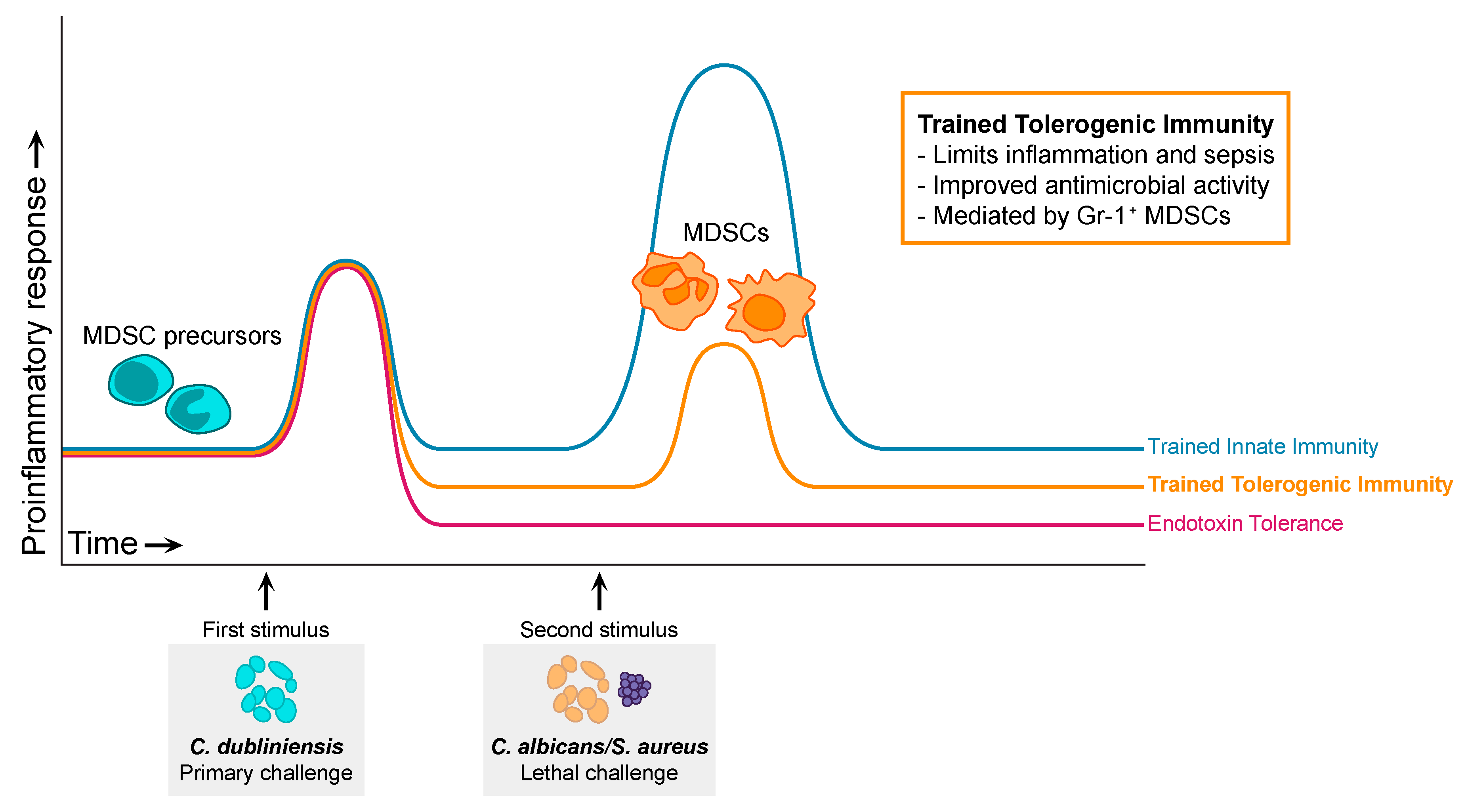
5.2. Trained Tolerogenic Immunity
5.3. Mechanisms of MDSC Protection in Sepsis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef]
- Vergidis, P.; Clancy, C.J.; Shields, R.K.; Park, S.Y.; Wildfeuer, B.N.; Simmons, R.L.; Nguyen, M.H. Intra-Abdominal Candidiasis: The Importance of Early Source Control and Antifungal Treatment. PLoS ONE 2016, 11, e0153247. [Google Scholar] [CrossRef]
- de Ruiter, J.; Weel, J.; Manusama, E.; Kingma, W.P.; van der Voort, P.H.J. The Epidemiology of Intra-Abdominal Flora in Critically Ill Patients with Secondary and Tertiary Abdominal Sepsis. Infection 2009, 37, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.G.; Serufo, J.C.; Silva, R.A.P.; Marra, B.A.; Reis, C.M.F.; Hamdan, J.S.; Nicoli, J.R.; Carvalho, M.A.R.; Farias, L.M. Microbiologic profile of intra-abdominal infections at Belo Horizonte, Brazil. Am. J. Infect. Control 2003, 31, 135–143. [Google Scholar] [CrossRef]
- Calandra, T.; Bille, J.; Schneider, R.; Mosimann, F.; Francioli, P. Clinical Significance of Candida Isolated from Peritoneum in Surgical Patients. Lancet 1989, 334, 1437–1440. [Google Scholar] [CrossRef]
- Montravers, P.; Gauzit, R.; Muller, C.; Marmuse, J.P.; Fichelle, A.; Desmonts, J.M. Emergence of Antibiotic-Resistant Bacteria in Cases of Peritonitis After Intraabdominal Surgery Affects the Efficacy of Empirical Antimicrobial Therapy. Clin. Infect. Dis. 1996, 23, 486–494. [Google Scholar] [CrossRef]
- Dupont, H.; Paugam-Burtz, C.; Muller-Serieys, C.; Fierobe, L.; Chosidow, D.; Marmuse, J.-P.; Mantz, J.; Desmonts, J.-M. Predictive factors of mortality due to polymicrobial peritonitis with Candida isolation in peritoneal fluid in critically III patients. Arch. Surg. 2002, 137, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.G.; Chong, T.W.; Smith, R.L.; Evans, H.L.; Pruett, T.L.; Sawyer, R.G. Comparison of fungal and nonfungal infections in a broad-based surgical patient population. Surg. Infect. 2005, 6, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Miles, R.; Hawley, C.M.; McDonald, S.P.; Brown, F.G.; Rosman, J.B.; Wiggins, K.J.; Bannister, K.M.; Johnson, D.W. Predictors and outcomes of fungal peritonitis in peritoneal dialysis patients. Kidney Int. 2009, 76, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Hasibeder, W.; Halabi, M. Candida peritonitis. Minerva Anestesiol. 2014, 80, 470–481. [Google Scholar]
- Perlroth, J.; Choi, B.; Spellberg, B. Nosocomial fungal infections: Epidemiology, diagnosis, and treatment. Med. Mycol. 2007, 45, 321–346. [Google Scholar] [CrossRef]
- Montravers, P.; Dupont, H.; Gauzit, R.; Veber, B.; Auboyer, C.; Blin, P.; Hennequin, C.; Martin, C. Candida as a risk factor for mortality in peritonitis. Crit. Care Med. 2006, 34, 646–652. [Google Scholar] [CrossRef]
- Blot, S.I.; Vandewoude, K.H.; De Waele, J.J. Candida peritonitis. Curr. Opin. Crit. Care 2007, 13, 195–199. [Google Scholar] [CrossRef]
- Yamabayashi, H. A zymosan-like substance extracted from Candida albicans. Med. J. Osaka Univ. 1958, 9, 11–21. [Google Scholar]
- Mankiewicz, E.; Liivak, M. Effect of Candida albicans on the Evolution of Experimental Tuberculosis. Nature 1960, 187, 250–251. [Google Scholar] [CrossRef]
- Carlson, E. Synergistic effect of Candida albicans and Staphylococcus aureus on mouse mortality. Infect. Immun. 1982, 38, 921–924. [Google Scholar]
- Carlson, E. Enhancement by Candida albicans of Staphylococcus aureus, Serratia marcescens, and Streptococcus faecalis in the establishment of infection in mice. Infect. Immun. 1983, 39, 193–197. [Google Scholar]
- Klaerner, H.G.; Uknis, M.E.; Acton, R.D.; Dahlberg, P.S.; Carlone-Jambor, C.; Dunn, D.L. Candida albicans and Escherichia coli are synergistic pathogens during experimental microbial peritonitis. J. Surg. Res. 1997, 70, 161–165. [Google Scholar] [CrossRef]
- Sawyer, R.G.; Adams, R.B.; May, A.K.; Rosenlof, L.K.; Pruett, T.L. Development of Candida albicans and C. albicans/Escherichia coli/Bacteroides fragilis intraperitoneal abscess models with demonstration of fungus-induced bacterial translocation. Med. Mycol. 1995, 33, 49–52. [Google Scholar] [CrossRef]
- Peters, B.M.; Noverr, M.C. Candida albicans-Staphylococcus aureus Polymicrobial Peritonitis Modulates Host Innate Immunity. Infect. Immun. 2013, 81, 2178–2189. [Google Scholar] [CrossRef]
- Nash, E.E.; Peters, B.M.; Fidel, P.L.; Noverr, M.C. Morphology-independent virulence of Candida species during polymicrobial intra-abdominal infections with Staphylococcus aureus. Infect. Immun. 2015, 84, 90–98. [Google Scholar] [CrossRef]
- Nash, E.E.; Peters, B.M.; Palmer, G.E.; Fidel, P.L.; Noverr, M.C. Morphogenesis Is Not Required for Candida albicans-Staphylococcus aureus Intra-Abdominal Infection-Mediated Dissemination and Lethal Sepsis. Infect. Immun. 2014, 82, 3426–3435. [Google Scholar] [CrossRef]
- Lilly, E.; Ikeh, M.; Nash, E.E.; Fidel, P.L.J.; Noverr, M.C. Immune Protection against Lethal Fungal-Bacterial Intra-Abdominal Infections. MBio 2018, 9, e01472-17. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; Van Der Meer, J.W.M. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Quintin, J.; Cheng, S.C.; van der Meer, J.W.; Netea, M.G. Innate immune memory: Towards a better understanding of host defense mechanisms. Curr. Opin. Immunol. 2014, 29, 1–7. [Google Scholar] [CrossRef]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Peranzoni, E.; Zilio, S.; Marigo, I.; Dolcetti, L.; Zanovello, P.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010. [Google Scholar] [CrossRef]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Cuenca, A.; Delano, M.; Kelly-Scumpia, K.M.; Moreno, C.; Scumpia, P.O.; LaFace, D.M.; Heyworth, P.G.; Efron, P.A.; Moldawer, L.L. A Paradoxical Role for Myeloid-Derived Suppressor Cells in Sepsis and Trauma. Mol. Med. 2011, 17, 1. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2016, 8, 3649–3665. [Google Scholar] [CrossRef]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.L.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 2007, 204, 1463–1474. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-Dose Granulocyte-Macrophage Colony-Stimulating Factor-Producing Vaccines Impair the Immune Response through the Recruitment of Myeloid Suppressor Cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef]
- Sawanobori, Y.; Ueha, S.; Kurachi, M.; Shimaoka, T.; Talmadge, J.E.; Abe, J.; Shono, Y.; Kitabatake, M.; Kakimi, K.; Mukaida, N.; et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008, 111, 5457–5466. [Google Scholar] [CrossRef]
- Menetrier-Caux, C.; Montmain, G.; Dieu, M.; Bain, C.; Favrot, M.; Caux, C.; Blay, J. Inhibition of the Differentiation of Dendritic Cells From CD34+ Progenitors by Tumor Cells: Role of Interleukin-6 and Macrophage Colony-Stimulating Factor. Blood 1998, 92, 4778–4791. [Google Scholar]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo. Blood 1998, 92, 4150–4166. [Google Scholar]
- Pan, P.-Y.; Wang, G.X.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.M.; Chen, S.-H. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008, 111, 219–228. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 Promotes Tumor Progression by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 62, 2343–2346. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Hood, K.C.; Zobel, L.C.; Shafer, L.R.; Coles, M.; Toth, B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int. Immunopharmacol. 2007, 7, 140–151. [Google Scholar] [CrossRef]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of Dendritic Cell Differentiation and Antitumor Immune Response in Cancer by Pharmacologic-Selective Inhibition of the Janus-Activated Kinase 2/Signal Transducers and Activators of Transcription 3 Pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G.; et al. IL-4-Induced Arginase 1 Suppresses Alloreactive T Cells in Tumor-Bearing Mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef]
- Highfill, S.L.; Rodriguez, P.C.; Zhou, Q.; Goetz, C.A.; Koehn, B.H.; Veenstra, R.; Taylor, P.A.; Panoskaltsis-Mortari, A.; Serody, J.S.; Munn, D.H.; et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood 2010, 116, 5738–5747. [Google Scholar] [CrossRef]
- Bunt, S.K.; Clements, V.K.; Hanson, E.M.; Sinha, P.; Ostrand-Rosenberg, S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 2009, 85, 996–1004. [Google Scholar] [CrossRef]
- Hong, E.-H.; Chang, S.-Y.; Lee, B.-R.; Kim, Y.-S.; Lee, J.-M.; Kang, C.-Y.; Kweon, M.-N.; Ko, H.-J. Blockade of Myd88 signaling induces antitumor effects by skewing the immunosuppressive function of myeloid-derived suppressor cells. Int. J. Cancer 2013, 132, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Brand, A.; Hector, A.; Graepler-Mainka, U.; Ost, M.; Schafer, I.; Wecker, I.; Neri, D.; Wirth, A.; Mays, L.; et al. Flagellin Induces Myeloid-Derived Suppressor Cells: Implications for Pseudomonas aeruginosa Infection in Cystic Fibrosis Lung Disease. J. Immunol. 2013, 190, 1276–1284. [Google Scholar] [CrossRef]
- Maruyama, A.; Shime, H.; Takeda, Y.; Azuma, M.; Matsumoto, M.; Seya, T. Pam2 lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochem. Biophys. Res. Commun. 2015, 457, 445–450. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Popovic, P.J.; Zeh, H.J.; Ochoa, J.B. Arginine and Immunity. J. Nutr. 2007, 137, 1681S–1686S. [Google Scholar] [CrossRef]
- Brito, C.; Naviliat, M.; Tiscornia, A.C.; Vuillier, F.; Gualco, G.; Dighiero, G.; Radi, R.; Cayota, A.M. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J. Immunol. 1999, 162, 3356–3366. [Google Scholar] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef]
- Lai, D.; Qin, C.; Shu, Q. Myeloid-Derived Suppressor Cells in Sepsis. Biomed Res. Int. 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mandruzzato, S.; Brandau, S.; Britten, C.M.; Bronte, V.; Damuzzo, V.; Gouttefangeas, C.; Maurer, D.; Ottensmeier, C.; van der Burg, S.H.; Welters, M.J.P.; et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: Results from an interim study. Cancer Immunol. Immunother. 2016, 65, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Ost, M.; Singh, A.; Peschel, A.; Mehling, R.; Rieber, N.; Hartl, D. Myeloid-Derived Suppressor Cells in Bacterial Infections. Front. Cell. Infect. Microbiol. 2016. [Google Scholar] [CrossRef]
- Schrijver, I.T.; Théroude, C.; Roger, T. Myeloid-Derived Suppressor Cells in Sepsis. Front. Immunol. 2019, 10, 327. [Google Scholar] [CrossRef]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Kozel, J.A.; Holzapfel, M.; Muirhead, D.E.; Kielian, T. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J. Immunol. 2014, 192, 3778–3792. [Google Scholar] [CrossRef]
- Skabytska, Y.; Wölbing, F.; Günther, C.; Köberle, M.; Kaesler, S.; Chen, K.-M.; Guenova, E.; Demircioglu, D.; Kempf, W.E.; Volz, T.; et al. Cutaneous Innate Immune Sensing of Toll-like Receptor 2-6 Ligands Suppresses T Cell Immunity by Inducing Myeloid-Derived Suppressor Cells. Immunity 2014, 41, 762–775. [Google Scholar] [CrossRef]
- Tebartz, C.; Horst, S.A.; Sparwasser, T.; Huehn, J.; Beineke, A.; Peters, G.; Medina, E. A Major Role for Myeloid-Derived Suppressor Cells and a Minor Role for Regulatory T Cells in Immunosuppression during Staphylococcus aureus Infection. J. Immunol. 2015, 194, 1100–1111. [Google Scholar] [CrossRef]
- du Plessis, N.; Loebenberg, L.; Kriel, M.; von Groote-Bidlingmaier, F.; Ribechini, E.; Loxton, A.G.; van Helden, P.D.; Lutz, M.B.; Walzl, G. Increased Frequency of Myeloid-derived Suppressor Cells during Active Tuberculosis and after Recent Mycobacterium tuberculosis Infection Suppresses T-Cell Function. Am. J. Respir. Crit. Care Med. 2013, 188, 724–732. [Google Scholar] [CrossRef]
- Knaul, J.K.; Jörg, S.; Oberbeck-Mueller, D.; Heinemann, E.; Scheuermann, L.; Brinkmann, V.; Mollenkopf, H.-J.; Yeremeev, V.; Kaufmann, S.H.E.; Dorhoi, A. Lung-Residing Myeloid-derived Suppressors Display Dual Functionality in Murine Pulmonary Tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Tsiganov, E.N.; Verbina, E.M.; Radaeva, T.V.; Sosunov, V.V.; Kosmiadi, G.A.; Nikitina, I.Y.; Lyadova, I.V. Gr-1(dim)CD11b+ Immature Myeloid-Derived Suppressor Cells but Not Neutrophils Are Markers of Lethal Tuberculosis Infection in Mice. J. Immunol. 2014, 192, 4718–4727. [Google Scholar] [CrossRef]
- Yang, B.; Wang, X.; Jiang, J.; Zhai, F.; Cheng, X. Identification of CD244-expressing myeloid-derived suppressor cells in patients with active tuberculosis. Immunol. Lett. 2014, 158, 66–72. [Google Scholar] [CrossRef] [PubMed]
- El Daker, S.; Sacchi, A.; Tempestilli, M.; Carducci, C.; Goletti, D.; Vanini, V.; Colizzi, V.; Lauria, F.N.; Martini, F.; Martino, A. Granulocytic myeloid derived suppressor cells expansion during active pulmonary tuberculosis is associated with high nitric oxide plasma level. PLoS ONE 2015, 10, e0123772. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Singh, A.; Öz, H.; Carevic, M.; Bouzani, M.; Amich, J.; Ost, M.; Ye, Z.; Ballbach, M.; Schäfer, I.; et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe 2015, 17, 507–514. [Google Scholar] [CrossRef]
- Uhel, F.; Azzaoui, I.; Grégoire, M.; Pangault, C.; Dulong, J.; Tadié, J.-M.; Gacouin, A.; Camus, C.; Cynober, L.; Fest, T.; et al. Early Expansion of Circulating Granulocytic Myeloid-derived Suppressor Cells Predicts Development of Nosocomial Infections in Patients with Sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 315–327. [Google Scholar] [CrossRef]
- Mathias, B.; Delmas, A.L.; Ozrazgat-Baslanti, T.; Vanzant, E.L.; Szpila, B.E.; Mohr, A.M.; Moore, F.A.; Brakenridge, S.C.; Brumback, B.A.; Moldawer, L.L.; et al. Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann. Surg. 2017, 265, 827–834. [Google Scholar] [CrossRef]
- Noel, G.; Wang, Q.; Osterburg, A.; Schwemberger, S.; James, L.; Haar, L.; Giacalone, N.; Thomas, I.; Ogle, C. A ribonucleotide reductase inhibitor reverses burn-induced inflammatory defects. Shock 2010, 34, 535–544. [Google Scholar] [CrossRef]
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010, 207, 1453–1464. [Google Scholar] [CrossRef]
- Derive, M.; Bouazza, Y.; Alauzet, C.; Gibot, S. Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med. 2012, 38, 1040–1049. [Google Scholar] [CrossRef]
- Brudecki, L.; Ferguson, D.A.; McCall, C.E.; El Gazzar, M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun. 2012, 80, 2026–2034. [Google Scholar] [CrossRef]
- Singh, A.; Lelis, F.; Braig, S.; Schäfer, I.; Hartl, D.; Rieber, N. Differential Regulation of Myeloid-Derived Suppressor Cells by Candida species. Front. Microbiol. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Lilly, E.A.; Yano, J.; Esher, S.K.; Hardie, E.; Fidel, P.L.J.; Noverr, M.C. Spectrum of Trained Innate Immunity Induced by Low Virulence Candida Species Against Lethal Polymicrobial Intra-Abdominal Infection. Infect. Immun. 2019, in press. [Google Scholar]
- Yáñez, A.; Murciano, C.; O’Connor, J.E.; Gozalbo, D.; Gil, M.L. Candida albicans triggers proliferation and differentiation of hematopoietic stem and progenitor cells by a MyD88-dependent signaling. Microbes Infect. 2009, 11, 531–535. [Google Scholar] [CrossRef]
- Yáñez, A.; Gil, M.L.; Murciano, C.; Gozalbo, D.; O’Connor, J.-E.; Flores, A. Signalling through TLR2/MyD88 induces differentiation of murine bone marrow stem and progenitor cells to functional phagocytes in response to Candida albicans. Cell. Microbiol. 2009, 12, 114–128. [Google Scholar]
- Yáñez, A.; Megías, J.; O’Connor, J.E.; Gozalbo, D.; Gil, M.L. Candida albicans induces selective development of macrophages and monocyte derived dendritic cells by a TLR2 dependent signalling. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Zhang, P.; Nelson, S.; Bagby, G.J.; Siggins, R.; Shellito, J.E.; Welsh, D.A. The Lineage−c-Kit+Sca-1+ Cell Response to Escherichia coli Bacteremia in Balb/c Mice. Stem Cells 2008, 26, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Oh, Y.K.; Kim, Y.J.; Youn, J.; Ahn, M.I. Escherichia coli up-regulates proinflammatory cytokine expression in granulocyte/macrophage lineages of CD34+ stem cells via p50 homodimeric NF-κB. Clin. Exp. Immunol. 2004, 137, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, L.M.; Quinton, L.J.; Bagby, G.J.; Nelson, S.; Wang, G.; Zhang, P. Escherichia coli pneumonia enhances granulopoiesis and the mobilization of myeloid progenitor cells into the systemic circulation. Crit. Care Med. 2004, 32, 1740–1746. [Google Scholar] [CrossRef]
- Rodriguez, S.; Chora, A.; Goumnerov, B.; Mumaw, C.; Goebel, W.S.; Fernandez, L.; Baydoun, H.; HogenEsch, H.; Dombkowski, D.M.; Karlewicz, C.A.; et al. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood 2009, 114, 4064–4076. [Google Scholar] [CrossRef] [PubMed]
- MacNamara, K.C.; Racine, R.; Chatterjee, M.; Borjesson, D.; Winslow, G.M. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect. Immun. 2009, 77, 4061–4069. [Google Scholar] [CrossRef]
- Johns, J.L.; MacNamara, K.C.; Walker, N.J.; Winslow, G.M.; Borjesson, D.L. Infection with Anaplasma phagocytophilum induces multilineage alterations in hematopoietic progenitor cells and peripheral blood cells. Infect. Immun. 2009, 77, 4070–4080. [Google Scholar] [CrossRef]
- Serbina, N.V.; Hohl, T.M.; Cherny, M.; Pamer, E.G. Selective Expansion of the Monocytic Lineage Directed by Bacterial Infection. J. Immunol. 2009, 183, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef]
- Scumpia, P.O.; Kelly-Scumpia, K.M.; Delano, M.J.; Weinstein, J.S.; Cuenca, A.G.; Al-Quran, S.; Bovio, I.; Akira, S.; Kumagai, Y.; Moldawer, L.L. Cutting edge: Bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J. Immunol. 2010, 184, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Editorial: The intricacy of choice: Can bacteria decide what type of myeloid cells to stimulate? J. Leukoc. Biol. 2014, 96, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, F.; Vecchiarelli, A.; Cenci, E.; Puccetti, P.; Marconi, P.; Cassone, A. Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infect. Immun. 1986, 51, 668–674. [Google Scholar] [PubMed]
- Bistoni, F.; Verducci, G.; Perito, S.; Vecchiarelli, A.; Puccetti, P.; Marconi, P.; Cassone, A. Immunomodulation by a low-virulence, agerminative variant of Candida albicans. Further evidence for macrophage activation as one of the effector mechanisms of nonspecific anti-infectious protection. J. Med. Vet. Mycol. 1988, 26, 285–299. [Google Scholar] [CrossRef]
- van’t Wout, J.W.; Poell, R.; van Furth, R. The Role of BCG/PPD-Activated Macrophages in Resistance against Systemic Candidiasis in Mice. Scand. J. Immunol. 1992, 36, 713–720. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef]
- Pena, O.M.; Pistolic, J.; Raj, D.; Fjell, C.D.; Hancock, R.E.W. Endotoxin Tolerance Represents a Distinctive State of Alternative Polarization (M2) in Human Mononuclear Cells. J. Immunol. 2011, 186, 7243–7254. [Google Scholar] [CrossRef]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Lahni, P.M.; Denenberg, A.G.; Poynter, S.E.; Wong, H.R.; Cook, J.A.; Zingarelli, B. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock 2008, 30, 267–273. [Google Scholar] [CrossRef] [PubMed]
- del Fresno, C.; García-Rio, F.; Gómez-Piña, V.; Soares-Schanoski, A.; Fernández-Ruíz, I.; Jurado, T.; Kajiji, T.; Shu, C.; Marín, E.; del Arroyo, A.G.; et al. Potent Phagocytic Activity with Impaired Antigen Presentation Identifying Lipopolysaccharide-Tolerant Human Monocytes: Demonstration in Isolated Monocytes from Cystic Fibrosis Patients. J. Immunol. 2009, 151, 1637–1645. [Google Scholar]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef]
- Adib-Conquy, M.; Cavaillon, J.-M. Compensatory anti-inflammatory response syndrome. Thromb. Haemost. 2009, 101, 36–47. [Google Scholar]
- Álvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-trans-Retinoic Acid Eliminates Immature Myeloid Cells from Tumor-bearing Mice and Improves the Effect of Vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar]
- Kusmartsev, S.; Gabrilovich, D.I. STAT1 Signaling Regulates Tumor-Associated Macrophage-Mediated T Cell Deletion. J. Immunol. 2014, 174, 4880–4891. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esher, S.K.; Fidel, P.L., Jr.; Noverr, M.C. Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity. J. Fungi 2019, 5, 37. https://doi.org/10.3390/jof5020037
Esher SK, Fidel PL Jr., Noverr MC. Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity. Journal of Fungi. 2019; 5(2):37. https://doi.org/10.3390/jof5020037
Chicago/Turabian StyleEsher, Shannon K., Paul L. Fidel, Jr., and Mairi C. Noverr. 2019. "Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity" Journal of Fungi 5, no. 2: 37. https://doi.org/10.3390/jof5020037
APA StyleEsher, S. K., Fidel, P. L., Jr., & Noverr, M. C. (2019). Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity. Journal of Fungi, 5(2), 37. https://doi.org/10.3390/jof5020037