Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease


 ,
, 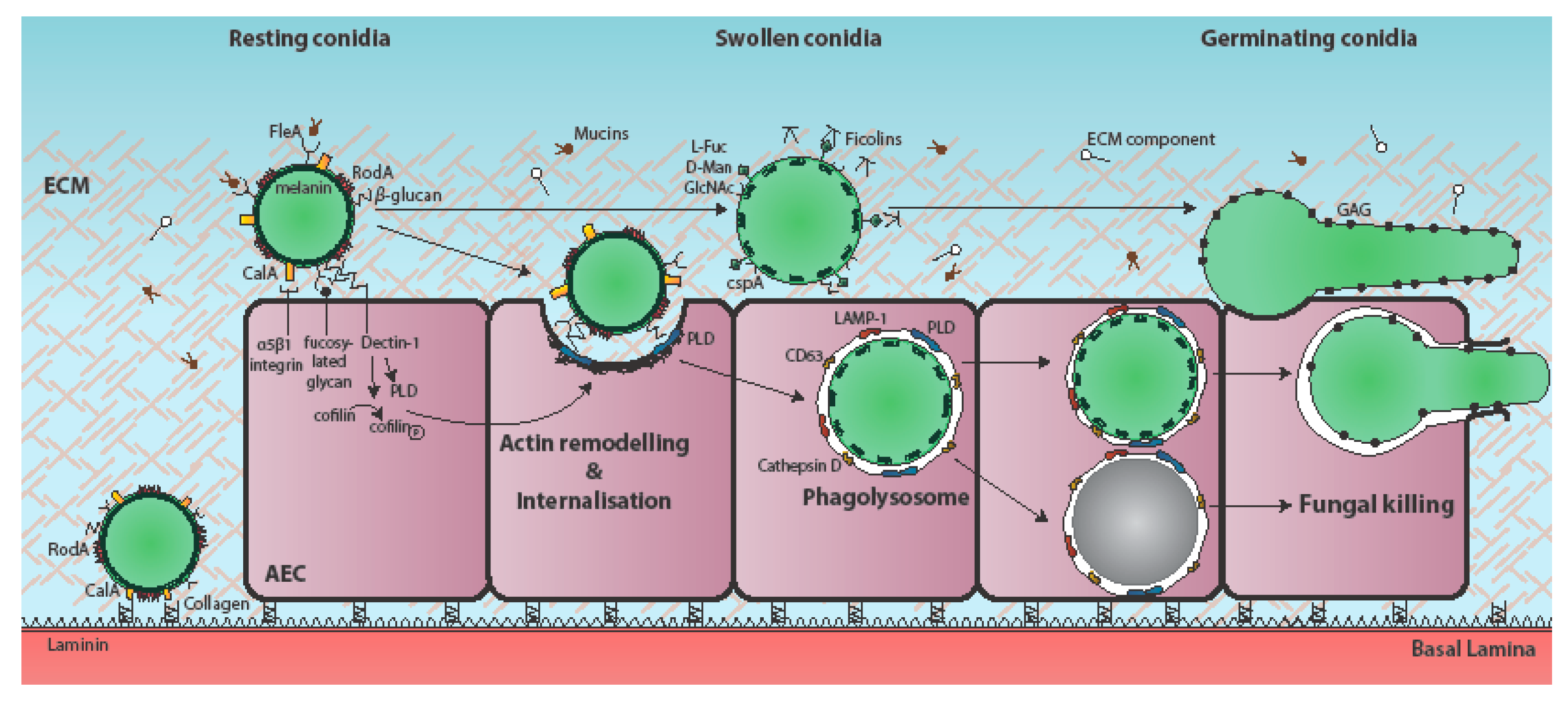{kind=link}
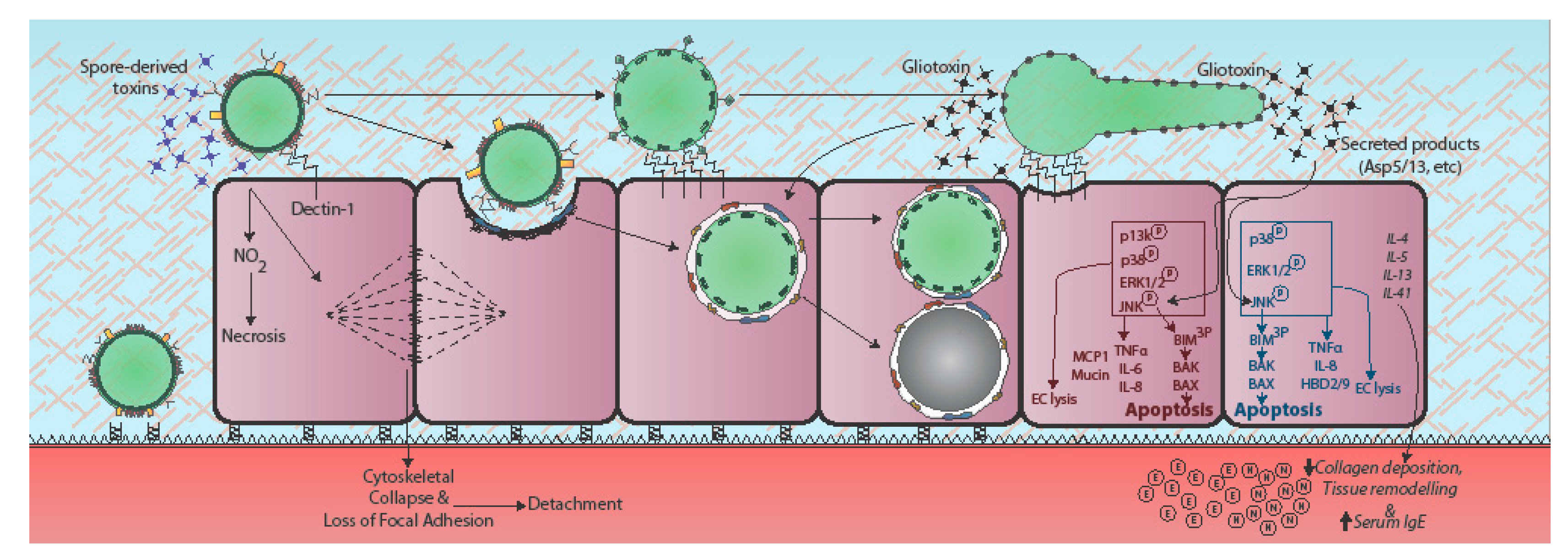{kind=link}
Abstract
1. Introduction
2. Attachment of A. fumigatus to the Respiratory Epithelium
3. Epithelial Uptake of A. fumigatus Spores
4. Epithelial Responses to A. fumigatus
5. Secreted Effectors of Epithelial Damage
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- Kleinkauf, N.; Verweij, P.; Arendrup, M.; Donnelly, P.; Cuenca-Estrella, M.; Fraaije, B.; Melchers, W.; Adriaenssens, N.; Kema, G.; Ullmann, A. Risk Assessment on the Impact of Environmental Usage of Triazoles on the Development and Spread of Resistance to Medical Triazoles in Aspergillus Species; European Centre for Disease Prevention and Control (ECDC): Stockholm, Sweden, 2013; ISBN 978-92-9193-444-7. [Google Scholar]
- Osherov, N. Interaction of the pathogenic mold Aspergillus fumigatus with lung epithelial cells. Front. Microbiol. 2012, 3, 346. [Google Scholar] [CrossRef] [PubMed]
- Croft, C.A.; Culibrk, L.; Moore, M.M.; Tebbutt, S.J. Interactions of Aspergillus fumigatus conidia with airway epithelial cells: A critical review. Front. Microbiol. 2016, 7, 472. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.C.; Filler, S.G. Host cell invasion by medically important fungi. Cold Spring Harb. Perspect. Med. 2015, 5, a019687. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J. Cell Sci. 2003, 116, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Uptake of Aspergillus fumigatus conidia by phagocytic and nonphagocytic cells in vitro: Quantitation using strains expressing green fluorescent protein. Infect. Immun. 2002, 70, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Munoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D.; et al. The pH-responsive pacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yu, R.; Zhen, D.; Tao, S.; Schmidt, M.; Han, L. β-1,3-glucan-induced host phospholipase D activation is involved in Aspergillus fumigatus internalization into type II human pneumocyte A549 cells. PLoS ONE 2011, 6, e21468. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Boisvieux-Ulrich, E.; Crestani, B.; Houcine, O.; Taramelli, D.; Lombardi, L.; Latgé, J.P. Internalization of Aspergillus fumigatus conidia by epithelial and endothelial cells. Infect. Immun. 1997, 65, 1510–1514. [Google Scholar] [PubMed]
- Zhang, Z.; Liu, R.; Noordhoek, J.A.; Kauffman, H.F. Interaction of airway epithelial cells (A549) with spores and mycelium of Aspergillus fumigatus. J. Infect. 2005, 51, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Gomez, P.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Functional genomics of human bronchial epithelial cells directly interacting with conidia of Aspergillus fumigatus. BMC Genom. 2010, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, J.L.; Gomez, P.; Ruan, J.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS ONE 2011, 6, e20527. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Datta, K.; Askin, F.B.; Staab, J.F.; Marr, K.A. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am. J. Respir. Crit. Care Med. 2012, 185, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Rammaert, B.; Jouvion, G.; de Chaumont, F.; Garcia-Hermoso, D.; Szczepaniak, C.; Renaudat, C.; Olivo-Marin, J.C.; Chretien, F.; Dromer, F.; Bretagne, S. Absence of fungal spore internalization by bronchial epithelium in mouse models evidenced by a new bioimaging approach and transmission electronic microscopy. Am. J. Pathol. 2015, 185, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Botterel, F.; Gross, K.; Ibrahim-Granet, O.; Khoufache, K.; Escabasse, V.; Coste, A.; Cordonnier, C.; Escudier, E.; Bretagne, S. Phagocytosis of Aspergillus fumigatus conidia by primary nasal epithelial cells in vitro. BMC Microbiol. 2008, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lee, M.J.; Solis, N.V.; Phan, Q.T.; Swidergall, M.; Ralph, B.; Ibrahim, A.S.; Sheppard, D.C.; Filler, S.G. Aspergillus fumigatus calA binds to integrin α5β1 and mediates host cell invasion. Nat. Microbiol. 2016, 2, 16211. [Google Scholar] [CrossRef] [PubMed]
- Levdansky, E.; Romano, J.; Shadkchan, Y.; Sharon, H.; Verstrepen, K.J.; Fink, G.R.; Osherov, N. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot. Cell 2007, 6, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Gravelat, F.N.; Beauvais, A.; Liu, H.; Lee, M.J.; Snarr, B.D.; Chen, D.; Xu, W.; Kravtsov, I.; Hoareau, C.M.; Vanier, G.; et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog. 2013, 9, e1003575. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Nakaguchi, A.; Sakai, K.; Takahashi, H.; Hagiwara, D.; Toyotome, T.; Chibana, H.; Watanabe, A.; Yaguchi, T.; Yamaguchi, M.; Kamei, K.; et al. Aspergillus fumigatus adhesion factors in dormant conidia revealed through comparative phenotypic and transcriptomic analyses. Cell. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bromley, I.M.; Donaldson, K. Binding of Aspergillus fumigatus spores to lung epithelial cells and basement membrane proteins: Relevance to the asthmatic lung. Thorax 1996, 51, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- DeHart, D.J.; Agwu, D.E.; Julian, N.C.; Washburn, R.G. Binding and germination of Aspergillus fumigatus conidia on cultured A549 pneumocytes. J. Infect. Dis. 1997, 175, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Wasylnka, J.A.; Moore, M.M. Adhesion of Aspergillus species to extracellular matrix proteins: Evidence for involvement of negatively charged carbohydrates on the conidial surface. Infect. Immun. 2000, 68, 3377–3384. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Debeaupuis, J.P.; Crameri, R.; Carey, M.; Charles, F.; Prevost, M.C.; Schmitt, C.; Philippe, B.; Latge, J.P. Conidial hydrophobins of Aspergillus fumigatus. Appl. Environ. Microbiol. 2003, 69, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Kuboi, S.; Ishimaru, T.; Tamada, S.; Bernard, E.M.; Perlin, D.S.; Armstrong, D. Molecular characterization of AfuFleA, an L-fucose-specific lectin from Aspergillus fumigatus. J. Infect. Chemother. 2013, 19, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Houser, J.; Komarek, J.; Kostlanova, N.; Cioci, G.; Varrot, A.; Kerr, S.C.; Lahmann, M.; Balloy, V.; Fahy, J.V.; Chignard, M.; et al. A soluble fucose-specific lectin from Aspergillus fumigatus conidia—Structure, specificity and possible role in fungal pathogenicity. PLoS ONE 2013, 8, e83077. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Lund, K.P.; Christensen, K.B.; Holm, A.T.; Dubey, L.K.; Moeller, J.B.; Jepsen, C.S.; Schlosser, A.; Galgoczy, L.; Thiel, S.; et al. M-ficolin is present in Aspergillus fumigatus infected lung and modulates epithelial cell immune responses elicited by fungal cell wall polysaccharides. Virulence 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Abdolrasouli, A.; Shah, A.; Reed, A.; Armstrong-James, D.; Schelenz, S. The serum opsonin L-ficolin is detected in lungs of human transplant recipients following fungal infections and modulates inflammation and killing of Aspergillus fumigatus. J. Infect. Dis. 2015, 212, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Bidula, S.; Sexton, D.W.; Yates, M.; Abdolrasouli, A.; Shah, A.; Wallis, R.; Reed, A.; Armstrong-James, D.; Schelenz, S. H-ficolin binds Aspergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology 2015, 146, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Han, J.; Yu, X. E-cadherin mediates adhesion of Aspergillus fumigatus to non-small cell lung cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Chen, F.; Sun, H.; Chen, C.; Zhao, B.L. Important factors mediates the adhesion of Aspergillus fumigatus to alveolar epithelial cells with E-cadherin. Am. J. Transl. Res. 2016, 8, 2419–2425. [Google Scholar] [PubMed]
- Bidula, S.; Kenawy, H.; Ali, Y.M.; Sexton, D.; Schwaeble, W.J.; Schelenz, S. Role of ficolin-A and lectin complement pathway in the innate defense against pathogenic Aspergillus species. Infect. Immun. 2013, 81, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Han, X.; Chen, F.; Jia, X.; Zhao, J.; Zhang, C.; Yong, C.; Tian, S.; Zhou, X.; Han, L. Evidence for the involvement of cofilin in Aspergillus fumigatus internalization into type II alveolar epithelial cells. BMC Microbiol. 2015, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Thywissen, A.; Heinekamp, T.; Saluz, H.P.; Brakhage, A.A. Melanin dependent survival of Aspergillus fumigatus conidia in lung epithelial cells. Int. J. Med. Microbiol. IJMM 2014, 304, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kogan, T.V.; Jadoun, J.; Mittelman, L.; Hirschberg, K.; Osherov, N. Involvement of secreted Aspergillus fumigatus proteases in disruption of the actin fiber cytoskeleton and loss of focal adhesion sites in infected A549 lung pneumocytes. J. Infect. Dis. 2004, 189, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.K.; Lu, X.; Li, X.; Sun, Q.Y.; Su, X.; Song, Y.; Sun, H.M.; Shi, Y. Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune responses in human bronchial epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Sallenave, J.M.; Wu, Y.; Touqui, L.; Latge, J.P.; Si-Tahar, M.; Chignard, M. Aspergillus fumigatus-induced interleukin-8 synthesis by respiratory epithelial cells is controlled by the phosphatidylinositol 3-kinase, p38 MAPK, and ERK1/2 pathways and not by the Toll-like receptor-myd88 pathway. J. Biol. Chem. 2008, 283, 30513–30521. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, L.; Huet, D.; Femenia, F.; Mouyna, I.; Abdelouahab, M.; Cagna, A.; Guerrier, D.; Tichanne-Seltzer, V.; Baeza-Squiban, A.; Chermette, R.; et al. Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol. 2009, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Sharon, H.; Amar, D.; Levdansky, E.; Mircus, G.; Shadkchan, Y.; Shamir, R.; Osherov, N. PrtT-regulated proteins secreted by Aspergillus fumigatus activate MAPK signaling in exposed A549 lung cells leading to necrotic cell death. PLoS ONE 2011, 6, e17509. [Google Scholar] [CrossRef] [PubMed]
- Icheoku, U.; Bertuzzi, M.; Thomson, D.; Moyes, D.L.; Bignell, E.M.; Naglik, J.R. Temporal analysis of the Aspergillus fumigatus host-pathogen interaction reveals morphotype-specific host activation and a dominant role for secreted fungal products in epithelial damage. J. Infect. Dis. 2018, submitted. [Google Scholar]
- Namvar, S.; Warn, P.; Farnell, E.; Bromley, M.; Fraczek, M.; Bowyer, P.; Herrick, S. Aspergillus fumigatus proteases, Asp f5 and Asp f13, are essential for airway inflammation and remodelling in a murine inhalation model. Clin. Exp. Allergy 2015, 45, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chen, F.; Pan, W.; Yu, R.; Tian, S.; Han, G.; Fang, H.; Wang, S.; Zhao, J.; Li, X.; et al. Gliotoxin promotes Aspergillus fumigatus internalization into type II human pneumocyte A549 cells by inducing host phospholipase d activation. Microbes Infect. 2014, 16, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Amitani, R.; Taylor, G.; Elezis, E.N.; Llewellyn-Jones, C.; Mitchell, J.; Kuze, F.; Cole, P.J.; Wilson, R. Purification and characterization of factors produced by Aspergillus fumigatus which affect human ciliated respiratory epithelium. Infect. Immun. 1995, 63, 3266–3271. [Google Scholar] [PubMed]
- Geissler, A.; Haun, F.; Frank, D.O.; Wieland, K.; Simon, M.M.; Idzko, M.; Davis, R.J.; Maurer, U.; Borner, C. Apoptosis induced by the fungal pathogen gliotoxin requires a triple phosphorylation of Bim by JNK. Cell Death Differ. 2013, 20, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zuo, J.; Han, Y.; Zhang, L.; Yan, L.; Shen, L.; Jiang, Y.; Cao, Y.; Zhao, J. Gliotoxin produced by Aspergillus fumigatus induces apoptosis of human bronchial epithelial cells via the Bcl-2 pathway. Int. J. Clin. Exp. Med. 2017, 10, 12. [Google Scholar]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Levdansky, E.; Kashi, O.; Sharon, H.; Shadkchan, Y.; Osherov, N. The Aspergillus fumigatus cspA gene encoding a repeat-rich cell wall protein is important for normal conidial cell wall architecture and interaction with host cells. Eukaryot. Cell 2010, 9, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.C.; Fischer, G.J.; Sinha, M.; McCabe, O.; Palmer, J.M.; Choera, T.; Yun Lim, F.; Wimmerova, M.; Carrington, S.D.; Yuan, S.; et al. FleA expression in Aspergillus fumigatus is recognized by fucosylated structures on mucins and macrophages to prevent lung infection. PLoS Pathog. 2016, 12, e1005555. [Google Scholar] [CrossRef] [PubMed]
- Gout, E.; Garlatti, V.; Smith, D.F.; Lacroix, M.; Dumestre-Pérard, C.; Lunardi, T.; Martin, L.; Cesbron, J.-Y.; Arlaud, G.J.; Gaboriaud, C.; et al. Carbohydrate recognition properties of human ficolins: Glycan array screening reveals the sialic acid binding specificity of M-ficolin. J. Biol. Chem. 2010, 285, 6612–6622. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Liu, Y.; Kanno, K.; Takahashi, M.; Matsushita, M.; Fujita, T. Identification of the mouse H-ficolin gene as a pseudogene and orthology between mouse ficolins a/b and human L-/M-ficolins. Genomics 2004, 84, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Genster, N.; Praestekjaer Cramer, E.; Rosbjerg, A.; Pilely, K.; Cowland, J.B.; Garred, P. Ficolins promote fungal clearance in vivo and modulate the inflammatory cytokine response in host defense against Aspergillus fumigatus. J. Innate Immun. 2016, 8, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, T.; Delangle, A.; Simenel, C.; Coddeville, B.; van Vliet, S.J.; van Kooyk, Y.; Bozza, S.; Moretti, S.; Schwarz, F.; Trichot, C.; et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Gravelat, F.N.; Cerone, R.P.; Baptista, S.D.; Campoli, P.V.; Choe, S.I.; Kravtsov, I.; Vinogradov, E.; Creuzenet, C.; Liu, H.; et al. Overlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J. Biol. Chem. 2014, 289, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Geller, A.M.; Bamford, N.C.; Liu, H.; Gravelat, F.N.; Snarr, B.D.; Le Mauff, F.; Chabot, J.; Ralph, B.; Ostapska, H.; et al. Deacetylation of fungal exopolysaccharide mediates adhesion and biofilm formation. mBio 2016, 7, e00252-16. [Google Scholar] [CrossRef] [PubMed]
- Escobar, N.; Ordonez, S.R.; Wösten, H.A.B.; Haas, P.-J.A.; de Cock, H.; Haagsman, H.P. Hide, keep quiet, and keep low: Properties that make Aspergillus fumigatus a successful lung pathogen. Front. Microbiol. 2016, 7, 438. [Google Scholar] [CrossRef] [PubMed]
- Belaish, R.; Sharon, H.; Levdansky, E.; Greenstein, S.; Shadkchan, Y.; Osherov, N. The Aspergillus nidulans cetA and calA genes are involved in conidial germination and cell wall morphogenesis. Fungal Genet. Biol. 2008, 45, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Mahajan, L.; Ramjee, S.; Singh, Y.; Basir, S.F.; Madan, T. Identification and characterization of a laminin-binding protein of Aspergillus fumigatus: Extracellular thaumatin domain protein (AfCalAp). J. Med. Microbiol. 2009, 58, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, C.; Jia, X.; Wang, S.; Wang, J.; Chen, Y.; Zhao, J.; Tian, S.; Han, X.; Han, L. Transcriptome profiles of human lung epithelial cells A549 interacting with Aspergillus fumigatus by RNA-seq. PLoS ONE 2015, 10, e0135720. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Di Ianni, M.; Bozza, S.; Giovannini, G.; Zagarella, S.; Zelante, T.; D’Angelo, C.; Pierini, A.; Pitzurra, L.; Falzetti, F.; et al. Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 2010, 116, 5394–5402. [Google Scholar] [CrossRef] [PubMed]
- Heyl, K.A.; Klassert, T.E.; Heinrich, A.; Muller, M.M.; Klaile, E.; Dienemann, H.; Grunewald, C.; Bals, R.; Singer, B.B.; Slevogt, H. Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable Haemophilus influenzae. mBio 2014, 5, e01492-14. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Yuk, J.M.; Shin, D.M.; Jo, E.K. Dectin-1 is inducible and plays an essential role for mycobacteria-induced innate immune responses in airway epithelial cells. J. Clin. Immunol. 2009, 29, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Jahn, B.; Boukhallouk, F.; Lotz, J.; Langfelder, K.; Wanner, G.; Brakhage, A.A. Interaction of human phagocytes with pigmentless Aspergillus conidia. Infect. Immun. 2000, 68, 3736–3739. [Google Scholar] [CrossRef] [PubMed]
- Volling, K.; Thywissen, A.; Brakhage, A.A.; Saluz, H.P. Phagocytosis of melanized aspergillus conidia by macrophages exerts cytoprotective effects by sustained PI3K/AKT signalling. Cell. Microbiol. 2011, 13, 1130–1148. [Google Scholar] [CrossRef] [PubMed]
- Thywißen, A.; Heinekamp, T.; Dahse, H.-M.; Schmaler-Ripcke, J.; Nietsche, S.; Zipfel, P.; Brakhage, A. Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front. Microbiol. 2011, 2, 96. [Google Scholar] [CrossRef] [PubMed]
- Chamilos, G.; Akoumianaki, T.; Kyrmizi, I.; Brakhage, A.; Beauvais, A.; Latge, J.-P. Melanin targets LC3-associated phagocytosis (LAP): A novel pathogenetic mechanism in fungal disease. Autophagy 2016, 12, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Akoumianaki, T.; Kyrmizi, I.; Valsecchi, I.; Gresnigt Mark, S.; Samonis, G.; Drakos, E.; Boumpas, D.; Muszkieta, L.; Prevost, M.-C.; Kontoyiannis Dimitrios, P.; et al. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe 2016, 19, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, C.; Hess, C.; Bals, R. Aspergillus fumigatus conidia induce interferon-beta signalling in respiratory epithelial cells. Eur. Respir. J. 2012, 39, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.; Wang, X.; Sifuentes Dos Santos, J.; Fysikopoulos, A.; Tadrist, S.; Canlet, C.; Artigot, M.P.; Loiseau, N.; Oswald, I.P.; Puel, O. Trypacidin, a spore-borne toxin from Aspergillus fumigatus, is cytotoxic to lung cells. PLoS ONE 2012, 7, e29906. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Wang, M.; Sun, W.K.; Xia, D.; Tan, M.M.; Ding, Y.; Qian, Q.; Su, X.; Shi, Y. Up-regulation of Dectin-1 in airway epithelial cells promotes mice defense against invasive pulmonary aspergillosis. Int. J. Clin. Exp. Med. 2015, 8, 17489–17497. [Google Scholar] [PubMed]
- Fekkar, A.; Balloy, V.; Pionneau, C.; Marinach-Patrice, C.; Chignard, M.; Mazier, D. Secretome of human bronchial epithelial cells in response to the fungal pathogen Aspergillus fumigatus analyzed by differential in-gel electrophoresis. J. Infect. Dis. 2012, 205, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Tomee, J.F.; Wierenga, A.T.; Hiemstra, P.S.; Kauffman, H.K. Proteases from Aspergillus fumigatus induce release of proinflammatory cytokines and cell detachment in airway epithelial cell lines. J. Infect. Dis. 1997, 176, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Seddigh, P.; Bracht, T.; Molinier-Frenkel, V.; Castellano, F.; Kniemeyer, O.; Schuster, M.; Weski, J.; Hasenberg, A.; Kraus, A.; Poschet, G.; et al. Quantitative analysis of proteome modulations in alveolar epithelial type II cells in response to pulmonary Aspergillus fumigatus infection. Mol. Cell. Proteom. 2017, 16, 2184–2198. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Huang, W.; Liang, J.; Guo, J.; Ji, J.; Yao, Y.; Zheng, M.; Cai, Z.; Lu, L.; Wang, J. IL4I1 is a novel regulator of M2 macrophage polarization that can inhibit T cell activation via L-tryptophan and arginine depletion and IL-10 production. PLoS ONE 2015, 10, e0142979. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.; Seyedmousavi, S.; Sugui, J.A.; Balenga, N.; Zhao, M.; Kwon Chung, K.J.; Biardel, S.; Laviolette, M.; Druey, K.M. Aspergillus fumigatus alkaline protease 1 (Alp1/Asp f13) in the airways correlates with asthma severity. J. Allergy Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Homma, T.; Norton, J.E.; Sha, Q.; Siebert, J.; Gupta, D.S.; Schroeder, J.W.; Schleimer, R.P. Suppression of epithelial signal transducer and activator of transcription 1 activation by extracts of Aspergillus fumigatus. Am. J. Respir. Cell Mol. Biol. 2015, 53, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Kato, A.; Bhushan, B.; Norton, J.E.; Suh, L.A.; Carter, R.G.; Gupta, D.S.; Schleimer, R.P. Role of Aspergillus fumigatus in triggering protease-activated receptor-2 in airway epithelial cells and skewing the cells toward a T-helper 2 bias. Am. J. Respir. Cell Mol. Biol. 2016, 54, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Dolan, S.K.; O’Keeffe, G.; Jones, G.W.; Doyle, S. Resistance is not futile: Gliotoxin biosynthesis, functionality and utility. Trends Microbiol. 2015, 23, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ami, R.; Lewis, R.E.; Leventakos, K.; Kontoyiannis, D.P. Aspergillus fumigatus inhibits angiogenesis through the production of gliotoxin and other secondary metabolites. Blood 2009, 114, 5393. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E.; Wiederhold, N.P.; Chi, J.; Han, X.Y.; Komanduri, K.V.; Kontoyiannis, D.P.; Prince, R.A. Detection of gliotoxin in experimental and human aspergillosis. Infect. Immun. 2005, 73, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Domingo, M.P.; Colmenarejo, C.; Martinez-Lostao, L.; Mullbacher, A.; Jarne, C.; Revillo, M.J.; Delgado, P.; Roc, L.; Meis, J.F.; Rezusta, A.; et al. Bis(methyl)gliotoxin proves to be a more stable and reliable marker for invasive aspergillosis than gliotoxin and suitable for use in diagnosis. Diagn. Microbiol. Infect. Dis. 2012, 73, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sugui, J.A.; Pardo, J.; Chang, Y.C.; Zarember, K.A.; Nardone, G.; Galvez, E.M.; Mullbacher, A.; Gallin, J.I.; Simon, M.M.; Kwon-Chung, K.J. Gliotoxin is a virulence factor of Aspergillus fumigatus: GliP deletion attenuates virulence in mice immunosuppressed with hydrocortisone. Eukaryot. Cell 2007, 6, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Spikes, S.; Xu, R.; Nguyen, C.K.; Chamilos, G.; Kontoyiannis, D.P.; Jacobson, R.H.; Ejzykowicz, D.E.; Chiang, L.Y.; Filler, S.G.; May, G.S. Gliotoxin production in Aspergillus fumigatus contributes to host-specific differences in virulence. J. Infect. Dis. 2008, 197, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Thorley, A.J.; Grandolfo, D.; Lim, E.; Goldstraw, P.; Young, A.; Tetley, T.D. Innate immune responses to bacterial ligands in the peripheral human lung--role of alveolar epithelial TLR expression and signalling. PLoS ONE 2011, 6, e21827. [Google Scholar] [CrossRef] [PubMed]
- Pollmächer, J.; Figge, M.T. Agent-based model of human alveoli predicts chemotactic signaling by epithelial cells during early Aspergillus fumigatus infection. PLoS ONE 2014, 9, e111630. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.J.; Boon, N.J.; Vrcelj, K.; Nguyen, A.; Vinci, C.; Armstrong-James, D.; Bignell, E. In silico modeling of spore inhalation reveals fungal persistence following low dose exposure. Sci. Rep. 2015, 5, 13958. [Google Scholar] [CrossRef] [PubMed]
- Seidler, M.J.; Salvenmoser, S.; Müller, F.-M.C. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob. Agents Chemother. 2008, 52, 4130–4136. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Bomme, P.; Lechner, B.E.; Mislin, G.L.A.; Lair, V.; Prévost, M.-C.; Latgé, J.-P.; Haas, H.; Beauvais, A. Pseudomonas aeruginosa manipulates redox and iron homeostasis of its microbiota partner Aspergillus fumigatus via phenazines. Sci. Rep. 2015, 5, 8220. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuzzi, M.; Hayes, G.E.; Icheoku, U.J.; Van Rhijn, N.; Denning, D.W.; Osherov, N.; Bignell, E.M. Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. J. Fungi 2018, 4, 8. https://doi.org/10.3390/jof4010008
Bertuzzi M, Hayes GE, Icheoku UJ, Van Rhijn N, Denning DW, Osherov N, Bignell EM. Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. Journal of Fungi. 2018; 4(1):8. https://doi.org/10.3390/jof4010008
Chicago/Turabian StyleBertuzzi, Margherita, Gemma E. Hayes, Uju J. Icheoku, Norman Van Rhijn, David W. Denning, Nir Osherov, and Elaine M. Bignell. 2018. "Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease" Journal of Fungi 4, no. 1: 8. https://doi.org/10.3390/jof4010008
APA StyleBertuzzi, M., Hayes, G. E., Icheoku, U. J., Van Rhijn, N., Denning, D. W., Osherov, N., & Bignell, E. M. (2018). Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. Journal of Fungi, 4(1), 8. https://doi.org/10.3390/jof4010008