
Diverse, Cryptic, and Undescribed: Club and Coral Fungi in a Temperate Australian Forest


Abstract
1. Introduction
2. Materials and Methods
2.1. Field Work and Sampling
2.2. DNA Extraction, PCR, and Specimen Identification
2.3. Reference Sequences
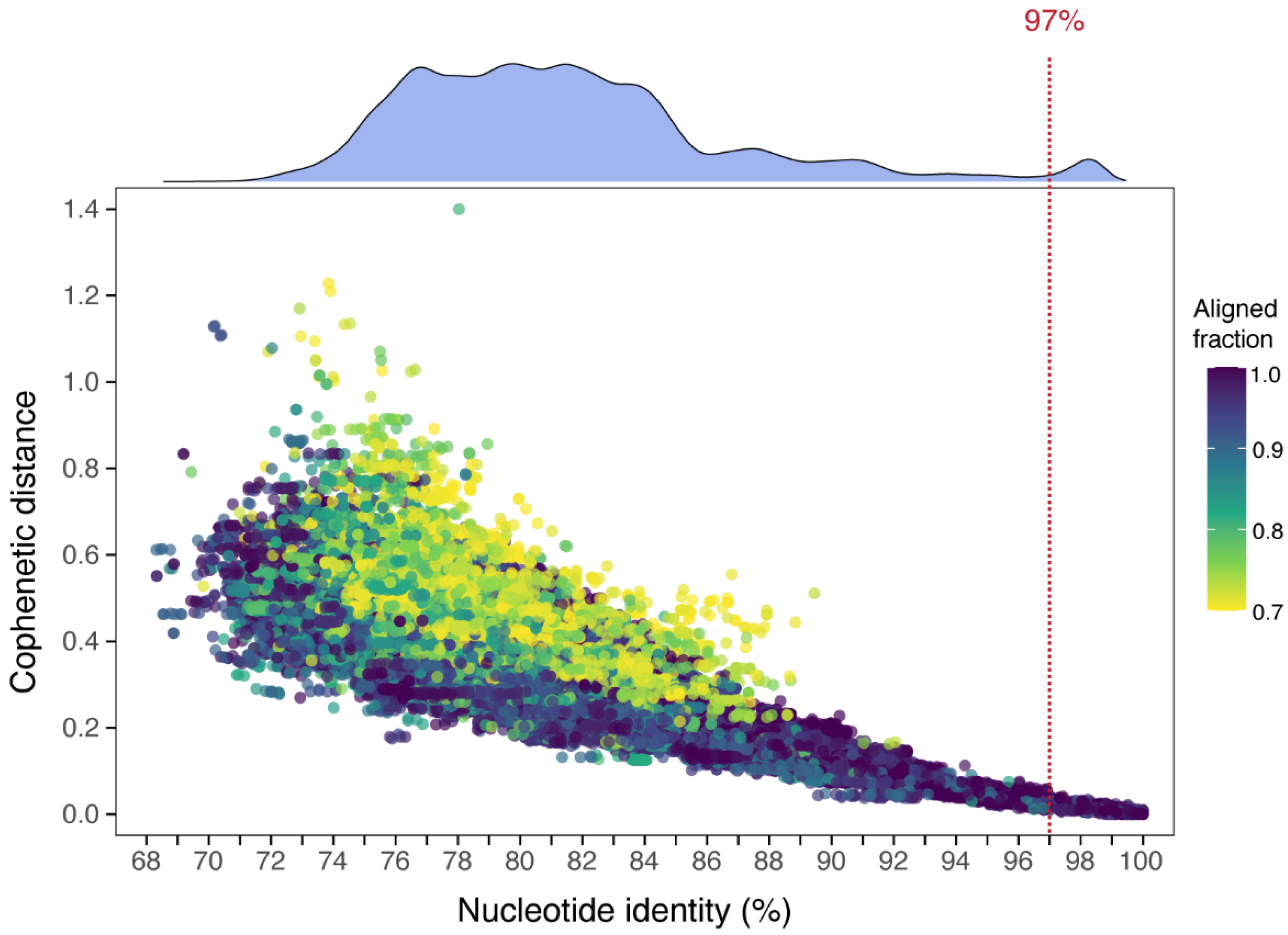
2.4. Establishing Criteria for Defining Operational Taxonomic Units (OTUs)
2.5. Assigning Sequences into OTUs
2.6. Genus Phylogenies
3. Results
3.1. Nucleotide Diversity and Definition of Operational Taxonomic Units
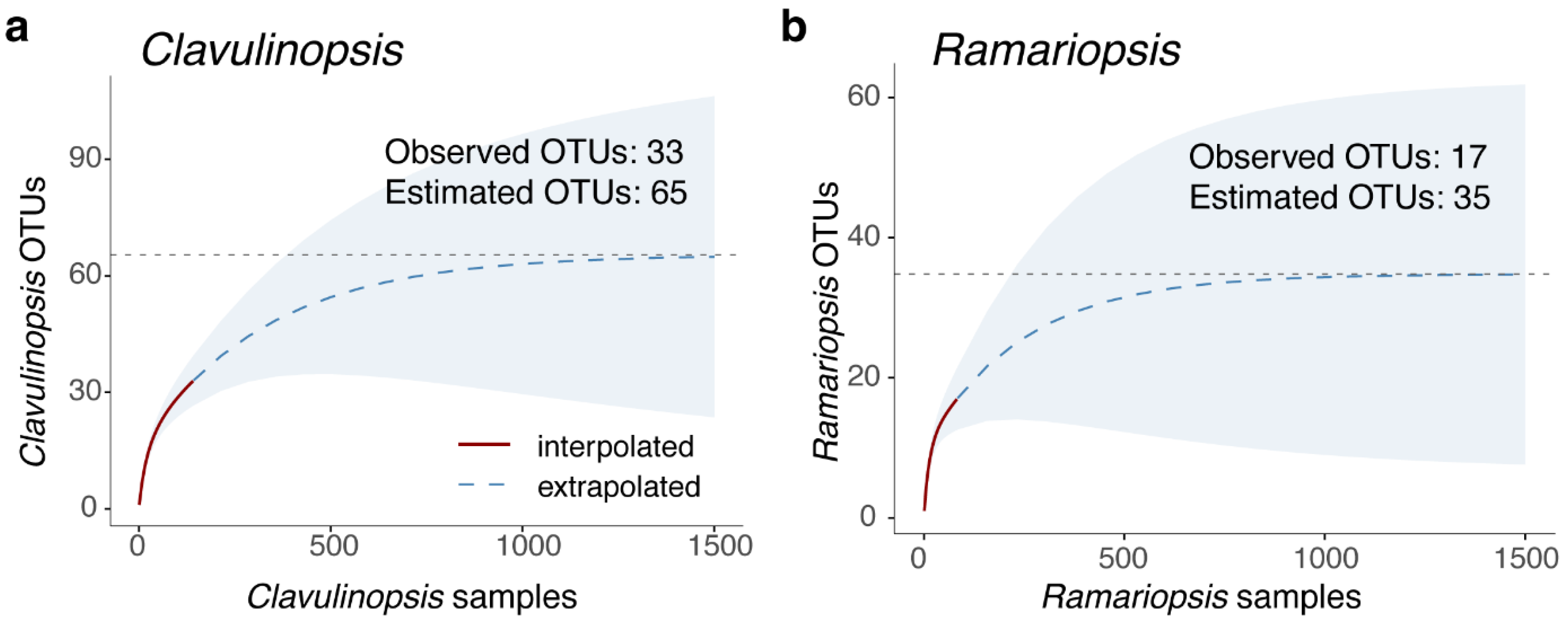
3.2. Clavulinopsis Diversity
3.3. Ramariopsis Diversity
3.4. Ramaria Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baldrian, P.; Větrovský, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2022, 114, 539–547. [Google Scholar] [CrossRef]
- Hawksworth, D.L. The magnitude of fungal diversity: The 1· 5 million species estimate revisited. Mycol. Res. 2001, 105, 1422–1432. [Google Scholar] [CrossRef]
- Hyde, K.D.; Saleh, A.; Aumentado, H.D.R.; Boekhout, T.; Bera, I.; Khyaju, S.; Bhunjun, C.S.; Chethana, K.W.T.; Phukhamsakda, C.; Doilom, M.; et al. Fungal numbers: Global needs for a realistic assessment. Fungal Divers. 2024, 128, 191–225. [Google Scholar] [CrossRef]
- Tedersoo, L.; Mikryukov, V.; Zizka, A.; Bahram, M.; Hagh-Doust, N.; Anslan, S.; Prylutskyi, O.; Delgado-Baquerizo, M.; Maestre, F.T.; Pärn, J.; et al. Global patterns in endemicity and vulnerability of soil fungi. Glob. Change Biol. 2022, 28, 6696–6710. [Google Scholar] [CrossRef]
- May, T.W. Documenting the fungal biodiversity of Australasia: From 1800 to 2000 and beyond. Aust. Syst. Bot. 2001, 14, 329–356. [Google Scholar] [CrossRef]
- Bahram, M.; Netherway, T.; Frioux, C.; Ferretti, P.; Coelho, L.P.; Geisen, S.; Bork, P.; Hildebrand, F. Metagenomic assessment of the global diversity and distribution of bacteria and fungi. Environ. Microbiol. 2021, 23, 316–326. [Google Scholar] [CrossRef]
- May, T.W.; Cooper, J.A.; Dahlberg, A.; Furci, G.; Minter, D.W.; Mueller, G.M.; Pouliot, A.; Yang, Z. Recognition of the discipline of conservation mycology. Conserv. Biol. 2019, 33, 733–736. [Google Scholar] [CrossRef]
- Irga, P.J.; Barker, K.; Torpy, F.R. Conservation mycology in Australia and the potential role of citizen science. Conserv. Biol. 2018, 32, 1031–1037. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef]
- Xu, J. Fungal DNA barcoding. Genome 2016, 59, 913–932. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B.; List, F.B.C.A.; Bolchacova, E.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Truong, C.; Mujic, A.B.; Healy, R.; Kuhar, F.; Furci, G.; Torres, D.; Niskanen, T.; Sandoval-Leiva, P.A.; Fernández, N.; Escobar, J.M.; et al. How to know the fungi: Combining field inventories and DNA-barcoding to document fungal diversity. New Phytol. 2017, 214, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D. The invisible dimension of fungal diversity. Science 2016, 351, 1150–1151. [Google Scholar] [CrossRef]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The fungal tree of life: From molecular systematics to genome-scale phylogenies. In The Fungal Kingdom; Wiley Online Library: New York, NY, USA, 2017; pp. 1–34. [Google Scholar]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Aoki, T.; Ariyawansa, H.A.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Fungal taxonomy and sequence-based nomenclature. Nat. Microbiol. 2021, 6, 540–548. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Thines, M.; Crous, P.W.; Aime, M.C.; Aoki, T.; Cai, L.; Hyde, K.D.; Miller, A.N.; Zhang, N.; Stadler, M. Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon. IMA Fungus 2018, 9, 177–183. [Google Scholar] [CrossRef]
- McPherson, V.J.; Gillings, M.M.; Gillings, M.R. Diversity and abundance of club and coral fungi in the Upper Lane Cove Valley. in Proceedings of the Linnean Society of New South Wales. Proc. Linn. Soc. New South Wales 2023, 145, 25–34. [Google Scholar]
- Lindsay, A.; Robinson, R.; May, T.; McMullan-Fisher, S.J.M. Guide to Surveying Fungi in Australia; Fungimap Inc.: South Yarra, Australia, 2013. [Google Scholar]
- Sunnucks, P.; Hales, D.F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 1996, 13, 510–524. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. A Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS 2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; Van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Zielezinski, A.; Gudyś, A.; Barylski, J.; Siminski, K.; Rozwalak, P.; Dutilh, B.E.; Deorowicz, S. Ultrafast and accurate sequence alignment and clustering of viral genomes. Nat. Methods 2025, 22, 1191–1194. [Google Scholar] [CrossRef]
- Hsieh, T.; Ma, K.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (H ill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package (2.5-7). Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists, 2020. Available online: https://sortie-admin.readyhosting.com/lme/R%20Packages/vegan.pdf (accessed on 1 February 2025).
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. ggtreeExtra: Compact visualization of richly annotated phylogenetic data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Nilsson, R.H.; Verkley, G.J. dnabarcoder: An open-source software package for analyzing and predicting DNA sequence similarity cut-offs for fungal sequence identification. Mol. Ecol. Resour. 2022, 22, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sandoval, R.; De Luna, E.; Estrada, A.; Villegas, M. A phylogeny of Ramariopsis and allied taxa. Mycotaxon 2005, 94, 265–292. [Google Scholar]
- Birkebak, J.M.; Mayor, J.R.; Ryberg, K.M.; Matheny, P.B. A systematic, morphological and ecological overview of the Clavariaceae (Agaricales). Mycologia 2013, 105, 896–911. [Google Scholar] [CrossRef]
- Franchi, P.; Marchetti, M. I funghi clavarioidi in Italia. In 2021: Associazione Micologica Bresadola; A.M.B. Fondazione Centro Studi Micologici: Trento, Italy, 2021. [Google Scholar]
- Yan, J.; Wen, J.; Li, G.-W.; Wu, S.-W.; Zhang, P. Taxonomy and phylogenetic relationships of Clavulinopsis (Clavariaceae, Agaricales): Description of six new species and one newly recorded species from China. J. Fungi 2023, 9, 656. [Google Scholar] [CrossRef]
- Petersen, R.H. The clavarioid fungi of New Zealand. DSIR (Dep. Sci. Ind. Res.) Bull. 1988, 236, 1–170. [Google Scholar]
- Matheny, P.B.; Wang, Z.; Binder, M.; Curtis, J.M.; Lim, Y.W.; Nilsson, R.H.; Hughes, K.W.; Hofstetter, V.; Ammirati, J.F.; Schoch, C.L. Contributions of rpb2 and tef1 to the phylogeny of mushrooms and allies (Basidiomycota, Fungi). Mol. Phylogenetics Evol. 2007, 43, 430–451. [Google Scholar] [CrossRef]
- Stielow, J.B.; Levesque, C.A.; Seifert, K.A.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.G.J.M.V.; Verkley, G.J.M.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia-Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef]
- Young, A.; Fechner, N. Australian Coralloid Fungi I–Ramaria capitata. Mycologist 2007, 26, 37–50. [Google Scholar]
- Young, A. Australian coralloid fungi IV–Ramaria filicicola. Muelleria 2014, 33, 13–19. [Google Scholar] [CrossRef]
- Bahram, M.; Netherway, T. Fungi as mediators linking organisms and ecosystems. FEMS Microbiol. Rev. 2022, 46, fuab058. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, M. The Fungi: 1, 2, 3… 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10–1128. [Google Scholar] [CrossRef]
- Zhang, Y.; Mo, M.; Yang, L.; Mi, F.; Cao, Y.; Liu, C.; Tang, X.; Wang, P.; Xu, J. Exploring the species diversity of edible mushrooms in Yunnan, Southwestern China, by DNA barcoding. J. Fungi 2021, 7, 310. [Google Scholar] [CrossRef]
- Green, J.L.; Holmes, A.J.; Westoby, M.; Oliver, I.; Briscoe, D.; Dangerfield, M.; Gillings, M.; Beattie, A.J. Spatial scaling of microbial eukaryote diversity. Nature 2004, 432, 747–750. [Google Scholar] [CrossRef]
- Talbot, J.M.; Bruns, T.D.; Taylor, J.W.; Smith, D.P.; Branco, S.; Glassman, S.I.; Erlandson, S.; Vilgalys, R.; Liao, H.-L.; Smith, M.E.; et al. Endemism and functional convergence across the North American soil mycobiome. Proc. Natl. Acad. Sci. USA 2014, 111, 6341–6346. [Google Scholar] [CrossRef]
- Větrovský, T.; Kohout, P.; Kopecký, M.; Machac, A.; Man, M.; Bahnmann, B.D.; Brabcová, V.; Choi, J.; Meszárošová, L.; Human, Z.R.; et al. A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat. Commun. 2019, 10, 5142. [Google Scholar] [CrossRef]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B.H. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar] [CrossRef]
- Williams, J.; Read, C.F.; Norton, T.; Dovers, S.; Burgman, M.; Proctor, W.; Anderson, H. Australia State of the Environment Report 2001 (Biodiversity Theme Report); CSIRO Publishing on behalf of the Department of the Environment and Heritage Commonwealth of Australia: Canberra, Australia, 2001. [Google Scholar]
- Dickman, C. Biodiversity in Australia: An overview. In Global Biodiversity; Apple Academic Press: Palm Bay, FL, USA, 2018; pp. 513–556. [Google Scholar]
- Hawksworth, D.L.; Rossman, A.Y. Where are all the undescribed fungi? Phytopathology 1997, 87, 888–891. [Google Scholar] [CrossRef]
- Stefani, F.O.; Jones, R.H.; May, T.W. Concordance of seven gene genealogies compared to phenotypic data reveals multiple cryptic species in Australian dermocyboid Cortinarius (Agaricales). Mol. Phylogenetics Evol. 2014, 71, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Ekanayaka, A.H.; Karunarathna, S.C.; Tibpromma, S.; Dutta, A.K.; Tennakoon, D.S.; Karunarathna, A.; Chukeatirote, E.; Dai, D.-Q.; Stephenson, S.L.; Maharachchikumbura, S.S.N.; et al. Species evolution: Cryptic species and phenotypic noise with a particular focus on fungal systematics. Front. Cell. Infect. Microbiol. 2025, 15, 1497085. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Robbertse, B.; Robert, V.; Vu, D.; Cardinali, G.; Irinyi, L.; Meyer, W.; Nilsson, R.H.; Hughes, K.; Kirk, P.M.; et al. Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for Fungi. Database 2014, 2014, bau061. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Ryberg, M.; Kristiansson, E.; Abarenkov, K.; Larsson, K.-H.; Kõljalg, U.; Fairhead, C. Taxonomic reliability of DNA sequences in public sequence databases: A fungal perspective. PLoS ONE 2006, 1, e59. [Google Scholar] [CrossRef]
- Osmundson, T.W.; Robert, V.A.; Schoch, C.L.; Baker, L.J.; Smith, A.; Robich, G.; Mizzan, L.; Garbelotto, M.M.; Kolokotronis, S.-O. Filling gaps in biodiversity knowledge for macrofungi: Contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS ONE 2013, 8, e62419. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a Unified Paradigm for Sequence-Based Identification of Fungi; Wiley Online Library: New York, NY, USA, 2013. [Google Scholar]
- Tedersoo, L.; Abarenkov, K.; Nilsson, R.H.; Schüssler, A.; Grelet, G.-A.; Kohout, P.; Oja, J.; Bonito, G.M.; Veldre, V.; Jairus, T.; et al. Tidying up international nucleotide sequence databases: Ecological, geographical and sequence quality annotation of ITS sequences of mycorrhizal fungi. PLoS ONE 2011, 6, e24940. [Google Scholar] [CrossRef]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.-C.; Giraldo, A.; et al. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 2015, 53, 313–337. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Source A | #ITS Sequences B | #OTUs C | #Specimen OTUs Matched D | #Ref Species E | Refs with Multiple OTUs F | |
|---|---|---|---|---|---|---|---|
| Clavulinopsis | Field | 142 | 33 | 6 (18.2%) | |||
| NCBI | 110 | 46 | 25 | 7 (28.0%) | |||
| Ramariopsis | Field | 83 | 17 | 2 (11.8%) | |||
| NCBI | 33 | 20 | 14 | 3 (21.4%) | |||
| Ramaria | Field | 109 | 30 | 1 (3.33%) | |||
| NCBI | 237 | 102 | 89 | 16 (18.0%) | |||
| Total | 714 | 80 | 168 | 9 (11.25%) | 128 | 25 (20.3%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McPherson, V.J.; Gillings, M.R.; Ghaly, T.M. Diverse, Cryptic, and Undescribed: Club and Coral Fungi in a Temperate Australian Forest. J. Fungi 2025, 11, 502. https://doi.org/10.3390/jof11070502
McPherson VJ, Gillings MR, Ghaly TM. Diverse, Cryptic, and Undescribed: Club and Coral Fungi in a Temperate Australian Forest. Journal of Fungi. 2025; 11(7):502. https://doi.org/10.3390/jof11070502
Chicago/Turabian StyleMcPherson, Vanessa J., Michael R. Gillings, and Timothy M. Ghaly. 2025. "Diverse, Cryptic, and Undescribed: Club and Coral Fungi in a Temperate Australian Forest" Journal of Fungi 11, no. 7: 502. https://doi.org/10.3390/jof11070502
APA StyleMcPherson, V. J., Gillings, M. R., & Ghaly, T. M. (2025). Diverse, Cryptic, and Undescribed: Club and Coral Fungi in a Temperate Australian Forest. Journal of Fungi, 11(7), 502. https://doi.org/10.3390/jof11070502