
Comparative Genomics Reveals Ancient and Unique Pathogenicity Features in Australian Fusarium oxysporum f. sp. vasinfectum

 , , , ,
, , , ,  , , and
, , and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungi Isolate Collection
2.2. DNA Extraction and Quality Assessment
2.3. Genome Assembly and QC
2.4. Genome Annotation
2.5. Comparative Genomics and Gene Orthology Analysis
3. Results
3.1. Genome Assembly
3.2. Genome Annotation and Functional Characteristics
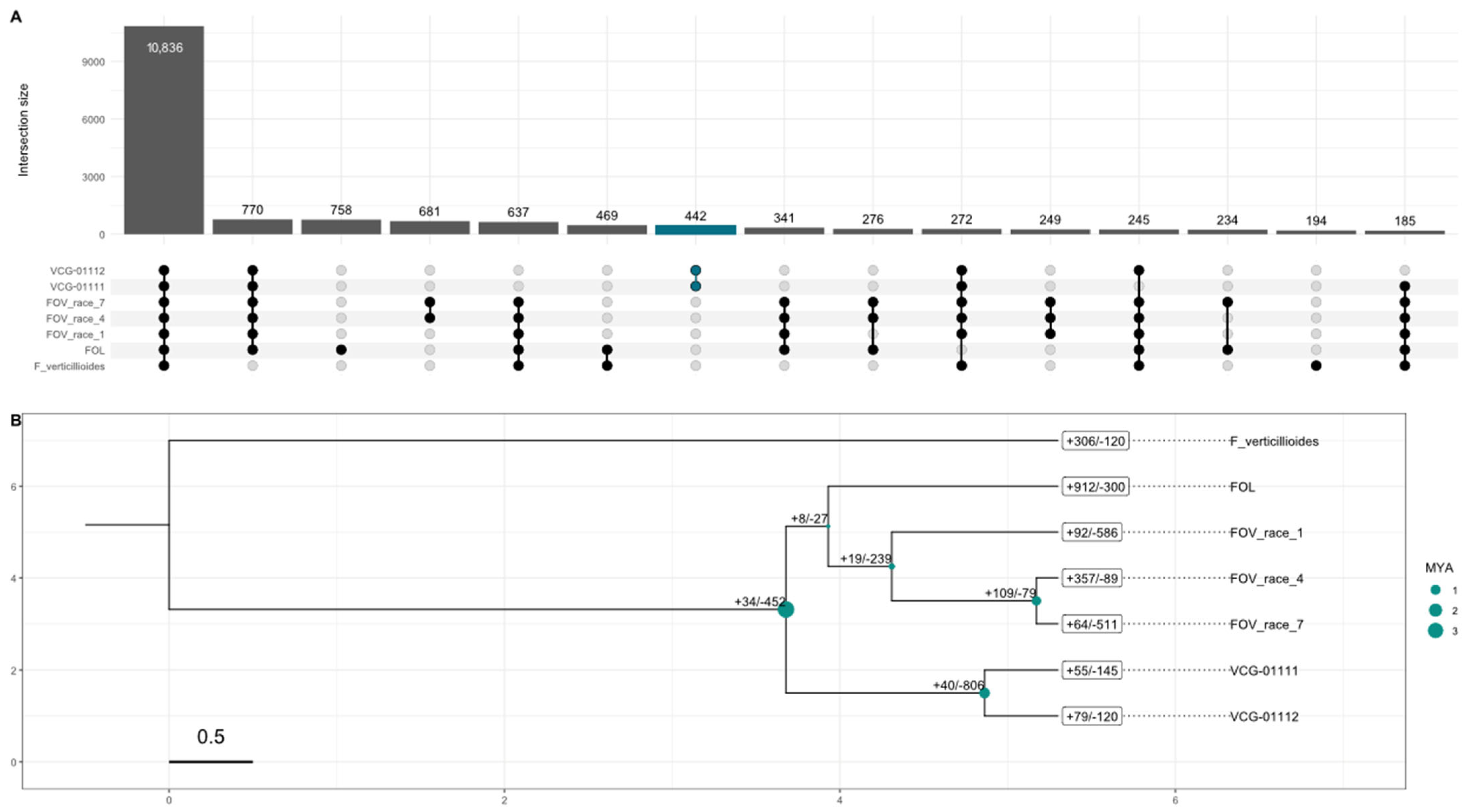
3.3. Evolutionary Differences and Gene Family Evolution
3.4. Identification of Secreted in Xylem Proteins (SIX)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, R.M.; Colyer, P.D.; Rothrock, C.S.; Kochman, J.K. Fusarium Wilt of Cotton: Population Diversity and Implications for Management. Plant Dis. 2006, 90, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.D.; Moore, N.Y.; Kochman, J.K. Characterisation of a Population of Fusarium oxysporum f.Sp. vasinfectum Causing Wilt of Cotton in Australia. Aust. J. Agric. Res. 1996, 47, 1143. [Google Scholar] [CrossRef]
- Chakrabarti, A. Fusarium oxysporum: A “Moving” View of Pathogenicity. In Soil Biology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 157–189. ISBN 978-3-642-39338-9. [Google Scholar]
- Bentley, S.; Kochman, J.K.; Moore, N.Y.; Pattemore, J.A.; Gulino, L.; O’Neill, W.T. DNA Diagnostics for Fusarium Wilt of Cotton; Australian Cotton Growers’ Research Association: Mungindi, Australia, 2000; pp. 455–461. [Google Scholar]
- Fernandez, D.; Assigbese, K.; Dubois, M.P.; Geiger, J.P. Molecular Characterization of Races and Vegetative Compatibility Groups in Fusarium oxysporum f. sp. vasinfectum. Appl. Environ. Microbiol. 1994, 60, 4039–4046. [Google Scholar] [CrossRef]
- Kim, Y.; Hutmacher, R.B.; Davis, R.M. Characterization of California Isolates of Fusarium oxysporum f. Sp. vasinfectum. Plant Dis. 2005, 89, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Le, D.P.; Nguyen, C.P.T.; Kafle, D.; Scheikowski, L.; Montgomery, J.; Lambeth, E.; Thomas, A.; O’Keeffe, K.; Shakeshaft, B.; Young, A.; et al. Surveillance, Diversity and Vegetative Compatibility Groups of Fusarium oxysporum f. Sp. vasinfectum Collected in Cotton Fields in Australia (2017 to 2022). Pathogens 2022, 11, 1537. [Google Scholar] [CrossRef]
- Le, D.P.; Tran, T.T.; Gregson, A.; Jackson, R. TEF1 Sequence-Based Diversity of Fusarium Species Recovered from Collar Rot Diseased Cotton Seedlings in New South Wales, Australia. Australas. Plant Pathol. 2020, 49, 277–284. [Google Scholar] [CrossRef]
- Wang, B.; Brubaker, C.L.; Summerell, B.A.; Thrall, P.H.; Burdon, J.J. Local Origin of Two Vegetative Compatibility Groups of Fusarium oxysporum f. Sp. vasinfectum in Australia: Evolutionary Origin of Cotton Wilt in Australia. Evol. Appl. 2010, 3, 505–524. [Google Scholar] [CrossRef]
- Craven, L.; Stewart, J.; Brown, A.; Grace, J. The Australian Wild Species of Gossypium. In Challenging the Future: Proceedings of the World Cotton Research Conference I, Brisbane, Australia, 14–17 February 1994; CSIRO: Pullenvale, Australia, 1995. [Google Scholar]
- Kochman, J. Fusarimn Wilt in Cotton—A New Record in Australia. Australas. Plant Pathol. 1995, 24, 74. [Google Scholar] [CrossRef]
- Stiller, W.N.; Wilson, I.W.; Stiller, W.N.; Wilson, I.W. Australian Cotton Germplasm Resources. In World Cotton Germplasm Resources; IntechOpen: London, UK, 2014; ISBN 978-953-51-1622-6. [Google Scholar]
- Conaty, W.C.; Broughton, K.J.; Egan, L.M.; Li, X.; Li, Z.; Liu, S.; Llewellyn, D.J.; MacMillan, C.P.; Moncuquet, P.; Rolland, V.; et al. Cotton Breeding in Australia: Meeting the Challenges of the 21st Century. Front. Plant Sci. 2022, 13, 904131. [Google Scholar] [CrossRef]
- McFadden, H.; Beasley, D.; Brubaker, C.L. Assessment of Gossypium sturtianum and G. Australe as Potential Sources of Fusarium Wilt Resistance to Cotton. Euphytica 2004, 138, 61–72. [Google Scholar] [CrossRef]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, S.; et al. Gene Family Expansions and Contractions Are Associated with Host Range in Plant Pathogens of the Genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef] [PubMed]
- Krijger, J.-J.; Thon, M.R.; Deising, H.B.; Wirsel, S.G. Compositions of Fungal Secretomes Indicate a Greater Impact of Phylogenetic History than Lifestyle Adaptation. BMC Genom. 2014, 15, 722. [Google Scholar] [CrossRef] [PubMed]
- Swett, C.L.; Del Castillo Múnera, J.; Hellman, E.; Helpio, E.; Gastelum, M.; Lopez Raymundo, E.; Johnson, H.; Oguchi, R.; Hopkins, A.; Beaulieu, J.; et al. Monitoring for a New I3 Resistance Gene-Breaking Race of F. oxysporum f. Sp. lycopersici (Fusarium Wilt) in California Processing Tomatoes Following Recent Widespread Adoption of Resistant (F3) Cultivars: Challenges with Race 3 and 4 Differentiation Methods. Front. Plant Sci. 2023, 14, 1088044. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Turra, D.; Zhou, S.; Ayhan, D.H.; DeIulio, G.A.; Guo, L.; Broz, K.; Wiederhold, N.; Coleman, J.J.; et al. The Genome of Opportunistic Fungal Pathogen Fusarium oxysporum Carries a Unique Set of Lineage-Specific Chromosomes. Commun. Biol. 2020, 3, 50. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Outram, M.A.; Smith, A.; McCombe, C.L.; Khambalkar, P.B.; Rima, S.A.; Sun, X.; Ma, L.; Ericsson, D.J.; Jones, D.A.; et al. The Structural Repertoire of Fusarium oxysporum f. Sp. lycopersici Effectors Revealed by Experimental and Computational Studies. eLife 2024, 12, RP89280. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, X.; Ming, Y.; Liu, C.; Zhang, X.; Liu, S.; Zhu, L. Characterization of the High-Quality Genome Sequence and Virulence Factors of Fusarium oxysporum f. Sp. vasinfectum Race 7. J. Fungi 2024, 10, 242. [Google Scholar] [CrossRef]
- Jobe, T.O.; Ulloa, M.; Ellis, M.L. Two de Novo Genome Assemblies from Pathogenic Fusarium oxysporum f. Sp. vasinfectum Race 4 (FOV4) Isolates from California. Microbiol. Resour. Announc. 2024, 13, e0076023. [Google Scholar] [CrossRef]
- Jobe, T.O.; Ulloa, M.; Ellis, M.L. A High-Quality Whole-Genome Sequence, Assembly, and Gene Annotation of Fusarium oxysporum f. Sp. vasinfectum (Fov) Race 1 from California. Microbiol. Resour. Announc. 2024, 13, e0070223. [Google Scholar] [CrossRef]
- Nash, S.M.; Snyder, W.C. Quantitative Estimations by Plate Counts of Propagules of the Bean Root Rot Fusarium in Field Soils. Phytopathology 1962, 52, 567–572. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid Isolation of High Molecular Weight Plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Schelkunov, M.I. Mabs, a Suite of Tools for Gene-Informed Genome Assembly. BMC Bioinform. 2023, 24, 377. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Rapid and Sensitive Detection of Genome Contamination at Scale with FCS-GX—PMC. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC10898089/ (accessed on 18 February 2025).
- Zhou, C.; McCarthy, S.A.; Durbin, R. YaHS: Yet Another Hi-C Scaffolding Tool. Bioinformatics 2023, 39, btac808. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ye, C.; Li, X.; Chen, Q.; Wu, Y.; Zhang, F.; Pan, R.; Zhang, S.; Chen, S.; Wang, X.; et al. quarTeT: A Telomere-to-Telomere Toolkit for Gap-Free Genome Assembly and Centromeric Repeat Identification. Hortic. Res. 2023, 10, uhad127. [Google Scholar] [CrossRef]
- Palmer, J.M.; Stajich, J. Funannotate v1.8.1: Eukaryotic Genome Annotation (v1.8.1). Zenodo. Available online: https://zenodo.org/records/4054262 (accessed on 19 June 2025).
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- InterProScan 5: Genome-Scale Protein Function Classification | Bioinformatics | Oxford Academic. Available online: https://academic.oup.com/bioinformatics/article/30/9/1236/237988?login=true (accessed on 18 February 2025).
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models | Nature Biotechnology. Available online: https://www.nature.com/articles/s41587-021-01156-3?utm_campaign=related_content&utm_source=HEALTH&utm_medium=communities (accessed on 18 February 2025).
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. biorXiv 2022. biorXiv:2022.04.08.487609. [Google Scholar]
- Jiang, Y.; Jiang, L.; Akhil, C.S.; Wang, D.; Zhang, Z.; Zhang, W.; Xu, D. MULocDeep Web Service for Protein Localization Prediction and Visualization at Subcellular and Suborganellar Levels. Nucleic Acids Res. 2023, 51, W343–W349. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An Integrated Platform for Exploring and Visualizing Orthologous Data across Genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics|Genome Biology | Full Text. Available online: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1832-y (accessed on 18 February 2025).
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Steenwyk, J.L.; LaBella, A.L.; Opulente, D.A.; Zhou, X.; Kominek, J.; Li, Y.; Groenewald, M.; Hittinger, C.T.; Rokas, A. Genome-Scale Phylogeny and Contrasting Modes of Genome Evolution in the Fungal Phylum Ascomycota. Sci. Adv. 2020, 6, eabd0079. [Google Scholar] [CrossRef] [PubMed]
- Da Lage, J.-L.; Binder, M.; Hua-Van, A.; Janeček, S.; Casane, D. Gene Make-up: Rapid and Massive Intron Gains after Horizontal Transfer of a Bacterial α-Amylase Gene to Basidiomycetes. BMC Evol. Biol. 2013, 13, 40. [Google Scholar] [CrossRef]
- Van der Nest, M.A.; Steenkamp, E.T.; McTaggart, A.R.; Trollip, C.; Godlonton, T.; Sauerman, E.; Roodt, D.; Naidoo, K.; Coetzee, M.P.A.; Wilken, P.M.; et al. Saprophytic and Pathogenic Fungi in the Ceratocystidaceae Differ in Their Ability to Metabolize Plant-Derived Sucrose. BMC Evol. Biol. 2015, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Rooney, A.P.; Proctor, R.H.; Brown, D.W.; McCormick, S.P.; Ward, T.J.; Frandsen, R.J.N.; Lysøe, E.; Rehner, S.A.; Aoki, T.; et al. Phylogenetic Analyses of RPB1 and RPB2 Support a Middle Cretaceous Origin for a Clade Comprising All Agriculturally and Medically Important Fusaria. Fungal Genet. Biol. 2013, 52, 20–31. [Google Scholar] [CrossRef]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot Plot Large Genomes in an Interactive, Efficient and Simple Way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Taxonomy and Evolution of the Cotton Genus, Gossypium—Wendel—2015—Agronomy Monographs—Wiley Online Library. Available online: https://acsess.onlinelibrary.wiley.com/doi/abs/10.2134/agronmonogr57.2013.0020 (accessed on 21 March 2025).
- Wagner, T.A.; Duke, S.E.; Davie, S.M.; Magill, C.; Liu, J. Interaction of Fusarium Wilt Race 4 with Root-Knot Nematode Increases Disease Severity in Cotton. Plant Dis. 2022, 106, 2558–2562. [Google Scholar] [CrossRef]
- Moore, N.Y.; Kochman, J.K.; Obst, N.R.; O’Neill, W.T.; Bentley, S. Fusarium Wilt of Cotton in Australia. In Proceedings of the World Cotton Research Conference—2, Athens, Greece, 6–12 September 1998. [Google Scholar]
- Chakrabarti, A.; Rep, M.; Wang, B.; Ashton, A.; Dodds, P.; Ellis, J. Variation in Potential Effector Genes Distinguishing Australian and Non-Australian Isolates of the Cotton Wilt Pathogen Fusarium oxysporum f.sp. vasinfectum. Plant Pathol. 2011, 60, 232–243. [Google Scholar] [CrossRef]
- Genetic Variation and Population Structure of Fusarium oxysporum f.sp. vasinfectum in Australia—Wang—2006—Plant Pathology—Wiley Online Library. Available online: https://bsppjournals.onlinelibrary.wiley.com/doi/full/10.1111/j.1365-3059.2006.01445.x (accessed on 18 February 2025).
- Sun, X.; Fang, X.; Wang, D.; Jones, D.A.; Ma, L. Transcriptome Analysis of Fusarium-Tomato Interaction Based on an Updated Genome Annotation of Fusarium oxysporum f. Sp. lycopersici Identifies Novel Effector Candidates That Suppress or Induce Cell Death in Nicotiana benthamiana. J. Fungi 2022, 8, 672. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lavalle, L.A.B.; Gillespie, V.J.; Tate, W.A.; Ellis, M.H.; Stiller, W.N.; Llewellyn, D.L.; Wilson, I.W. Molecular Mapping of a New Source of Fusarium Wilt Resistance in Tetraploid Cotton (Gossypium hirsutum L.). Mol. Breed. 2012, 30, 1181–1191. [Google Scholar] [CrossRef]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. [Fusarium oxysporum f. sp. Lycopersici] Genome Assembly GCF_000149955.1 (ASM14995v2). NCBI Datasets. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000149955.1/ (accessed on 12 November 2024).
- National Center for Biotechnology Information. [Fusarium verticillioides] Genome Assembly GCA_000149555.1 (ASM14955v1). NCBI Datasets. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000149555.1/ (accessed on 12 November 2024).
- Popa-Baez, A.; Smith, L.J.; Stiller, W.; Soliveres, M.; Pandey, G.; Wilson, I. The complete genome of two Australian Isolates of Fusarium oxysporum f. sp. vasinfectum. v1. CSIRO. Data Collect. 2025. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa-Baez, A.D.; Smith, L.J.; Stiller, W.N.; Soliveres, M.; Pandey, G.; Saski, C.A.; Jones, D.C.; Wilson, I.W. Comparative Genomics Reveals Ancient and Unique Pathogenicity Features in Australian Fusarium oxysporum f. sp. vasinfectum. J. Fungi 2025, 11, 481. https://doi.org/10.3390/jof11070481
Popa-Baez AD, Smith LJ, Stiller WN, Soliveres M, Pandey G, Saski CA, Jones DC, Wilson IW. Comparative Genomics Reveals Ancient and Unique Pathogenicity Features in Australian Fusarium oxysporum f. sp. vasinfectum. Journal of Fungi. 2025; 11(7):481. https://doi.org/10.3390/jof11070481
Chicago/Turabian StylePopa-Baez, Angel David, Linda J. Smith, Warwick N. Stiller, Melanie Soliveres, Gunjan Pandey, Christopher A. Saski, Don C. Jones, and Iain W. Wilson. 2025. "Comparative Genomics Reveals Ancient and Unique Pathogenicity Features in Australian Fusarium oxysporum f. sp. vasinfectum" Journal of Fungi 11, no. 7: 481. https://doi.org/10.3390/jof11070481
APA StylePopa-Baez, A. D., Smith, L. J., Stiller, W. N., Soliveres, M., Pandey, G., Saski, C. A., Jones, D. C., & Wilson, I. W. (2025). Comparative Genomics Reveals Ancient and Unique Pathogenicity Features in Australian Fusarium oxysporum f. sp. vasinfectum. Journal of Fungi, 11(7), 481. https://doi.org/10.3390/jof11070481