
Whole-Genome Characterization of Inonotus hispidus from Ulmus macrocarpa and Its Comparative Genomics with Strains from Morus alba and Acer truncatum


Abstract
1. Introduction
2. Materials and Methods
2.1. Screening and Obtaining Mononuclear Strains of UMI and AMI
2.2. Genomic DNA Extraction and Whole-Genome Sequencing of UMI and AMI
2.3. Genome Prediction and Functional Annotation
2.4. Comparative Genomic Analysis
2.5. Biological Characterization of UMI, AMI, and MAI
3. Results and Discussion
3.1. Identification of Mononuclear Strains from UMI and AMI
3.2. Genome Assembly and General Features of UMI
3.3. Genome-Wide Functional Annotation of UMI
3.3.1. General Functional Annotation Overview
3.3.2. KEGG Pathway Annotation and Core Metabolic Processes
3.3.3. CAZyme Families and Lignocellulose Degradation Potential
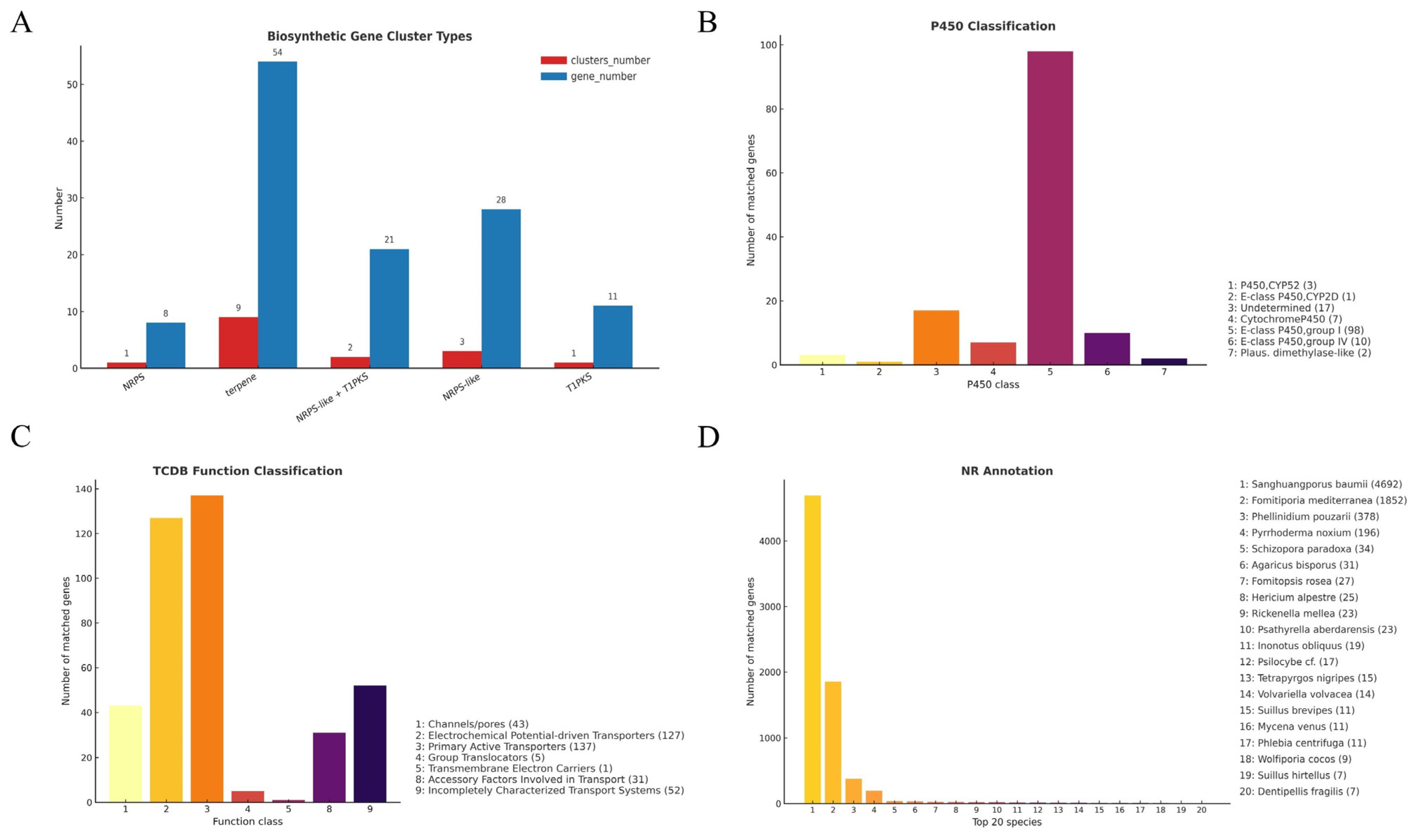
3.3.4. Cytochrome P450 Gene Family Analysis
3.3.5. Membrane Transporters Annotated via TCDB
3.3.6. Overall Annotation Coverage and Species-Level Homology
3.4. Comparative Genomic Analysis of UMI, AMI, and MAI Strains
3.4.1. Comparison Results of Whole-Genome Sequencing
3.4.2. Genome Collinearity and Rearrangement
3.4.3. Indel, SNP, and SV Analysis
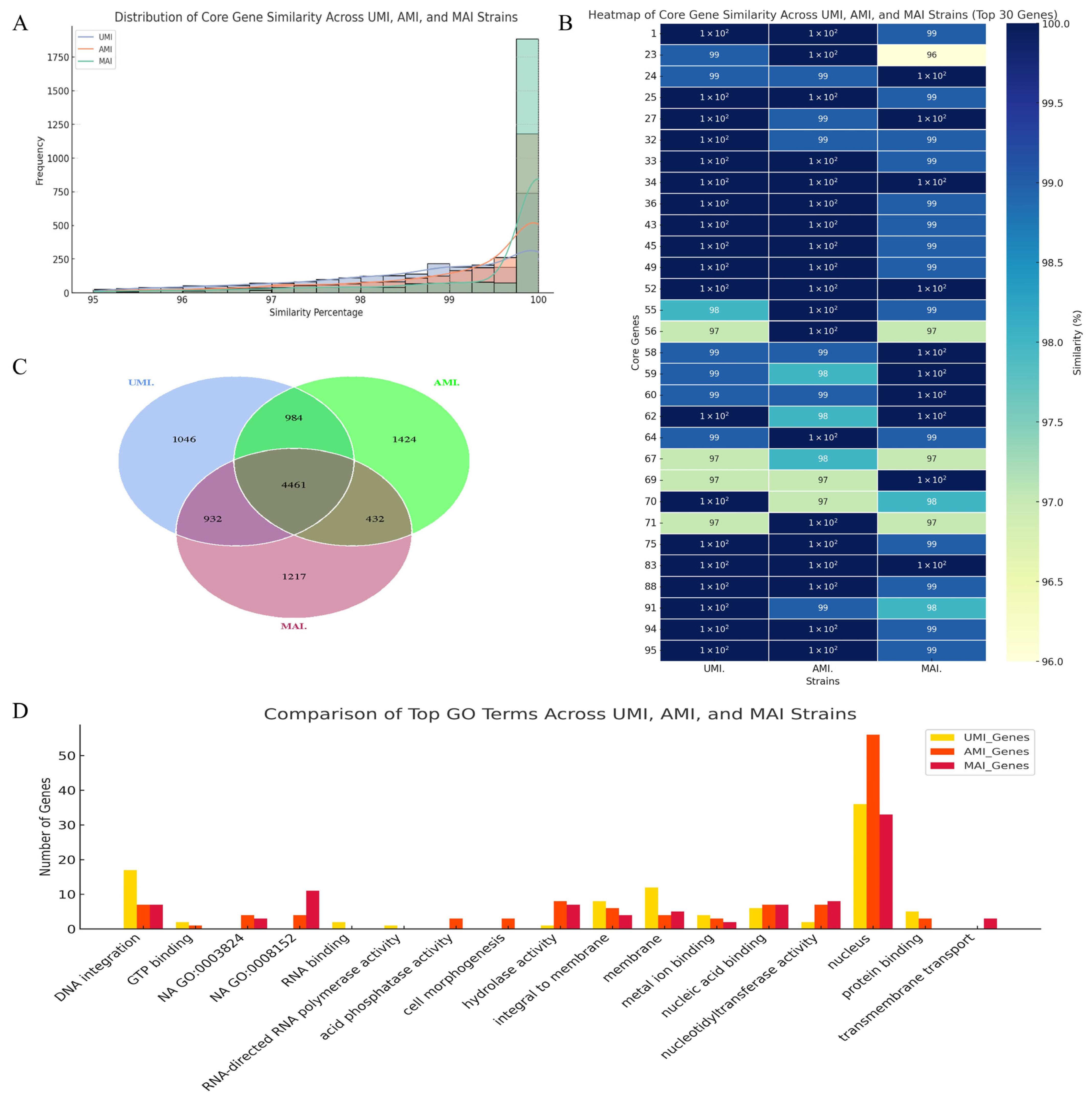
3.4.4. Core-Pan Genome Analysis
3.5. Comparative Analysis of Phenotypic Characteristics of UMI, AMI, and MAI Strains
3.6. Genomic Features and Application Potential of I. hispidus Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhou, L.W.; Yang, Z.L.; Bau, T.; Li, T.H.; Dai, Y.C. Resource diversity of Chinese macrofungi: Edible, medicinal and poisonous species. Fungal Divers. 2019, 98, 1–76. [Google Scholar] [CrossRef]
- Markakis, E.A.; Kavroulakis, N.; Ntougias, S.; Koubouris, G.C.; Sergentani, C.K.; Ligoxigakis, E.K. Characterization of fungi associated with wood decay of tree species and grapevine in Greece. Plant Dis. 2017, 11, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Feng, X.L.; Liu, C.; Gao, J.M.; Qi, J. Diverse metabolites and pharmacological effects from the basidiomycetes Inonotus hispidus. Antibiotics 2022, 8, 1097. [Google Scholar] [CrossRef]
- Yuan, Y.; Bian, L.S.; Wu, Y.D.; Chen, J.J.; Wu, F.; Liu, H.G.; Zeng, G.Y.; Dai, Y.C. Species diversity of pathogenic wood-rotting fungi (Agaricomycetes, Basidiomycota) in China. Mycology 2023, 3, 204–226. [Google Scholar] [CrossRef]
- Zhang, F.; Xue, F.; Xu, H.; Yuan, Y.; Wu, X.; Zhang, J.; Fu, J. Optimization of solid-state fermentation extraction of Inonotus hispidus fruiting body melanin. Foods 2021, 12, 2893. [Google Scholar] [CrossRef]
- Li, Z.; Bao, H. Anti-tumor effect of Inonotus hispidus petroleum ether extract in H22 tumor-bearing mice and analysis its mechanism by untargeted metabonomic. J. Ethnopharmacol. 2022, 285, 114898. [Google Scholar] [CrossRef]
- Liu, X.; Hou, R.; Yan, J.; Xu, K.; Wu, X.; Lin, W.; Zheng, M.; Fu, J. Purification and characterization of Inonotus hispidus exopolysaccharide and its protective effect on acute alcoholic liver injury in mice. Int. J. Biol. Macromol. 2019, 129, 41–49. [Google Scholar] [CrossRef]
- Wang, Q.; Bao, H.; Li, Z. Genomic comparison between two Inonotus hispidus strains isolated from growing in different tree species. Front. Genet. 2023, 14, 1221491. [Google Scholar] [CrossRef]
- Wang, Q.; Bao, H. Research on the mechanism of root endophytes of Morus alba L. and Fraxinus mandshurica Rupr., two host plants growing Inonotus hispidus (Bull.) P. Karst., with metabarcoding and metabolomics. Horticulturae 2024, 10, 1074. [Google Scholar] [CrossRef]
- Li, Z.; Bao, H. Comparative analysis of metabolic compositions and trace elements of Inonotus hispidus mushroom grown on five different tree species. ACS Omega 2022, 11, 9343–9358. [Google Scholar] [CrossRef]
- Han, C.; Bao, H. Therapeutic effects and mechanisms of Inonotus hispidus extract and active monomer compounds in a rat mammary gland hyperplasia model. J. Ethnopharmacol. 2024, 319 Pt 2, 117274. [Google Scholar] [CrossRef] [PubMed]
- Horbach, R.; Navarro-quesada, A.R.; Knogge, W.; Deising, H.B. When and how to kill a plant cell: Infection strategies of plant pathogenic fungi. J. Plant Physiol. 2011, 168, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 212, 4327–4341. [Google Scholar] [CrossRef]
- Jochen, K.; Fouts, D.E.; Shanmuga, S. Next generation sequencing technologies and the changing landscape of phage genomics. Bacteriophage 2012, 3, 190–199. [Google Scholar]
- Wu, T.; Cai, M.; Hu, H.; Jiao, C.; Zhang, Z.; Liu, Y.; Chen, J.; Xiao, C.; Li, X.; Gao, X.; et al. Whole-genome sequencing and transcriptome analysis of Ganoderma lucidum strain Yw-1-5 provides new insights into the enhanced effect of tween 80 on exopolysaccharide production. J. Fungi 2022, 10, 1081. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, Z.; Liu, Y.; Zhang, G.; Li, G. The whole-genome sequencing and analysis of a Ganoderma lucidum strain provide insights into the genetic basis of its high triterpene content. Genomics 2021, 113 Pt 2, 840–849. [Google Scholar] [CrossRef]
- Tang, S.; Jin, L.; Lei, P.; Shao, C.; Wu, S.; Yang, Y.; He, Y.; Ren, R.; Xu, J. Whole-genome assembly and analysis of a medicinal fungus: Inonotus hispidus. Front. Microbiol. 2022, 13, 967135. [Google Scholar] [CrossRef]
- Duan, M.; Bao, H.; Bau, T. Analyses of transcriptomes and the first complete genome of Leucocalocybe mongolica provide new insights into phylogenetic relationships and conservation. Sci. Rep. 2021, 11, 2930. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, F.; Si, J.; Zhao, Y.F.; Dai, Y.C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 2019, 111, 50–58. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, H.; Cai, M.; Liang, X.; Wu, X.; Wang, A.; Chen, X.; Li, X.; Xiao, C.; Huang, L.; et al. Whole genome sequencing of an edible and medicinal mushroom, Russula griseocarnosa, and its association with mycorrhizal characteristics. Gene 2022, 808, 145996. [Google Scholar] [CrossRef]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 3, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Berlin, K.; Koren, S.; Chin, C.S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 6, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 9, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Volpato, M.; Hull, M.; Carr, I.M. GO term viewer: Visualization of gene ontology enrichment in multiple differential gene expression analyses. Bioinform. Biol. Insights 2024, 18, 11779322241271550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 4, 1063–1070. [Google Scholar] [CrossRef]
- Kumari, S.; Magdaleno, J.S.L.; Grewal, R.K.; Rao, M.P.N.; Shaikh, A.R.; Cavallo, L.; Chawla, M.; Kumar, M. High potential for biomass-degrading CAZymes revealed by pine forest soil metagenomics. J. Biomol. Struct. Dyn. 2024, 21, 11483–11494. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signal P 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Aljouie, A.; Zhong, L.; Roshan, U. High scoring segment selection for pairwise whole genome sequence alignment with the maximum scoring subsequence and GPUs. Int. J. Comput. Biol. Drug Des. 2020, 13, 71. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2020, 7285, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Agnestisia, R.; Ono, A.; Nakamura, L.; Chino, R.; Nodera, K.; Aiso-Sanada, H.; Nezu, I.; Ishiguri, F.; Suzuki, T.; Yokota, S. The complete mitochondrial genome sequence of the medicinal fungus Inonotus obliquus (Hymenochaetaceae, Basidiomycota). Mitochondrial DNA B Resour. 2019, 2, 3504–3506. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.M.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef]
- Song, T.; Xu, F.; Shen, Y.; Fan, L.; Cai, W. The complete mitochondrial genome of Sanghuangporus vaninii Zhehuang-1 (Hymenochaetales, Basidiomycota). Mitochondrial DNA B Resour. 2021, 3, 1096–1097. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, S.; Zhou, L. The first whole genome sequencing of Sanghuangporus sanghuang provides insights into its medicinal application and evolution. J. Fungi 2021, 7, 787. [Google Scholar] [CrossRef]
- Wang, X.; Wei, J.; Liu, Z.; Wang, Y.; Yuan, X.; Wang, D.; Niu, J.; Yang, Y.; Zhou, J. Comparative genomic analysis of Sanghuangporus sanghuang with other Hymenochaetaceae species. Braz. J. Microbiol. 2024, 55, 87–100. [Google Scholar] [CrossRef]
- Chen, J.; Xu, J.; Wang, Q.; Ding, S.; Yang, R.; Zeng, W.; Wang, X. Comparative genomic analysis reveals lignocellulolytic enzymes and secondary metabolism genes in Lentinula edodes. Front. Microbiol. 2022, 13, 872911. [Google Scholar]
- Yang, Y.; Wang, Q.; Zhang, J.; Xia, X.; Wang, W.; Wang, P.; He, X. Genome and transcriptome analyses reveal high-yield triterpenoid biosynthesis in Ganoderma lucidum. Phytochemistry 2016, 131, 68–77. [Google Scholar]
- Zhang, R.; Feng, X.; Wang, Z.; Xie, T.; Duan, Y.; Liu, C.; Gao, J.; Qi, J. Genomic and metabolomic analyses of the medicinal fungus Inonotus hispidus. J. Fungi 2022, 8, 1245. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Fan, W.L.; Wang, W.F.; Chen, T.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 44, E4743–E4752. [Google Scholar]
- Min, B.; Kim, S.; Oh, Y.L.; Kong, W.S.; Park, H.; Cho, H.; Jang, K.Y.; Kim, J.G.; Choi, I.G. Genomic discovery of the hypsin gene and biosynthetic pathways for terpenoids in Hypsizygus marmoreus. BMC Genom. 2018, 1, 789. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, H.; Wang, Y.; Cheng, Y.; Li, Q.; Zhang, X.; Li, Y.; Liu, C.; Yang, R. Comparative genome and transcriptome analysis reveals evolutionary divergence and candidate genes involved in secondary metabolism in Inonotus obliquus. Front. Microbiol. 2021, 12, 678250. [Google Scholar]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity. BMC Biol. 2019, 17, 58. [Google Scholar] [CrossRef]
- Andrade, A.C.; Fróes, A.; Lopes, F.Á.C.; Thompson, F.L.; Krüger, R.H.; Dinsdale, E.; Bruce, T. Diversity of microbial carbohydrate-active enZYmes (CAZYmes) associated with freshwater and soil samples from Caatinga Biome. Microb. Ecol. 2017, 74, 89–105. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, X.; Yang, Y.L.; Xing, Y.M.; Zhang, Q.; Li, J.M.; Ma, K.; Liu, H.W.; Guo, S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017, 7, 10151. [Google Scholar] [CrossRef]
- Shim, D.; Park, S.G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.S.; Ro, H.S.; Kim, M.; Ryoo, R.; Rhee, S.K.; et al. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom. Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.; Xu, Y.; Wang, Y.; He, S.; Wu, B. Comparative genomics and lignocellulolytic enzyme evolution in wood-decaying fungi. Fungal Divers. 2023, 95, 1–22. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Floudas, D.; Held, B.W.; Riley, R.; Nagy, L.G.; Kovács, G.M.; Randlett, T.; Gácser, A.; Sipiczki, M.; Davis, J.M.; Doty, S.L.; et al. The evolution of lignin-degrading enzymes in Agaricomycetes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, C.; Zhang, L.; Xiao, Y. Genome mining and comparative analysis of cytochrome P450 gene families in medicinal fungi. Genes 2020, 11, 1524. [Google Scholar]
- Wu, B.; Xu, Z.; Knudsen, G.R.; Lin, F.; Zhang, B.; He, S. Comparative genomics of wood-decaying fungi reveals candidate genes for secondary metabolism and lignocellulose degradation. Fungal Genet. Biol. 2022, 159, 103666. [Google Scholar]
- Sonnenberg, A.S.M.; Gao, W.; Lavrijssen, B.; Hendrickx, P.M.; Sedaghat-Tellgerd, N.; Foulongne-Oriol, M.; Baars, J.J.P.; Visser, R.G.F.; Schaap, P.J.; Gehrmann, T.; et al. A detailed analysis of the whole genome of Agaricus bisporus reveals dynamic genome organization associated with mushroom formation and host interaction. Fungal Genet. Biol. 2016, 94, 39–53. [Google Scholar]
- Xiao, Y.; Cheng, X.; Liu, J.; Li, C.; Nong, W.; Bian, Y.; Cheung, M.K.; Kwan, H.S. Population genomic analysis uncovers environmental stress-driven selection and adaptation of Lentinula edodes population in China. Sci. Rep. 2016, 6, 36789. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, B.; Xu, Y.; Zhao, Q.; He, S. Comparative genomics reveals strain-specific CAZyme repertoires and metabolic adaptation in wood-decaying fungi. Fungal Biol. Rev. 2021, 35, 202–213. [Google Scholar]
- O’Connor, E.; Doyle, S.; Labarre, A.; Eastwood, D.C.; Burton, K.S. Proteomic investigation of interhyphal interactions between strains of Agaricus bisporus. Fungal Biol. 2020, 124, 792–804. [Google Scholar] [CrossRef]
- Moretti, S.; Losi, D.; Ghignone, S.; Onofri, S.; Voyron, S.; Pioli, S.; Gonthier, P. Characterization of non-enzymatic radical-generating mechanisms adopted by Fomitiporia mediterranea. J. Fungi 2023, 9, 498. [Google Scholar] [CrossRef]
- Sun, S.; Zhao, R.; Xu, J.; Ma, Y.; Shen, X.; Guo, L.; Liu, Y.; Zhu, C.; Cao, Y.; Zhang, J.; et al. Harnessing genotype-by-environment interactions in mushroom breeding: A systems biology perspective. Appl. Microbiol. Biotechnol. 2022, 106, 2601–2616. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | UMI |
|---|---|
| Genome size (bp) | 36,444,366 |
| Gene number | 9097 |
| Gene total length (bp) | 13,783,881 |
| Gene average length (bp) | 1515 |
| Gene length/genome (%) | 37.82 |
| Gene internal length | 22,660,485 |
| Gene internal GC content | 46.4 |
| tRNA | 111 |
| snRNA | 10 |
| LTR | 2519 |
| DNA | 555 |
| LINE | 392 |
| SINE | 11 |
| RC | 35 |
| TR | 3363 |
| Minisatellite DNA | 2279 |
| Microsatellite DNA | 615 |
| Gene ID | KO Term | Gene Name and Definition | EC Number |
|---|---|---|---|
| A2154 | hexPS, COQ1 | hexaprenyl-diphosphate synthase | 2.5.1.82 2.5.1.83 |
| A2770 | idi, IDI | isopentenyl-diphosphate Delta-isomerase | 5.3.3.2 |
| A2861 | PCYOX1, FCLY | prenylcysteine oxidase/farnesylcysteine lyase | 1.8.3.5 1.8.3.6 |
| A3529 | DHDDS, RER2, SRT1 | ditrans, polycis-polyprenyl diphosphate synthase | 2.5.1.87 |
| A3653 | HMGCR | hydroxymethylglutaryl-CoA reductase (NADPH) | 1.1.1.34 |
| A4362 | FDPS | farnesyl diphosphate synthase | 2.5.1.1 2.5.1.10 |
| A4588 | E2.3.3.10 | hydroxymethylglutaryl-CoA synthase | 2.3.3.10 |
| A5296 | MVD, mvaD | diphosphomevalonate decarboxylase | 4.1.1.33 |
| A5897 | E2.7.4.2, mvaK2 | phosphomevalonate kinase | 2.7.4.2 |
| A6225 | E2.3.1.9, atoB | acetyl-CoA C-acetyltransferase | 2.3.1.9 |
| A8924 | FNTA | protein farnesyltransferase/geranylgeranyltransferase type-1 subunit alpha | 2.5.1.58 2.5.1.59 |
| A0619 | STE24 | STE24 endopeptidase | 3.4.24.84 |
| A0708 | FNTB | protein farnesyltransferase subunit beta | 2.5.1.58 |
| Statistics | Sample | ||
|---|---|---|---|
| UMI | AMI | MAI | |
| Genome size (bp) | 36,444,366 | 33,809,908 | 34,136,716 |
| Gene number | 9097 | 8366 | 8356 |
| Gene total length (bp) | 13,783,881 | 11,802,855 | 12,839,852 |
| Gene average length (bp) | 1515 | 1411 | 1537 |
| Gene length/genome (%) | 37.82% | 34.91% | 37.61% |
| Genome size (bp) | 36,444,366 | 33,809,908 | 34,136,716 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, R.; Wang, Q.; Bao, H. Whole-Genome Characterization of Inonotus hispidus from Ulmus macrocarpa and Its Comparative Genomics with Strains from Morus alba and Acer truncatum. J. Fungi 2025, 11, 346. https://doi.org/10.3390/jof11050346
Bai R, Wang Q, Bao H. Whole-Genome Characterization of Inonotus hispidus from Ulmus macrocarpa and Its Comparative Genomics with Strains from Morus alba and Acer truncatum. Journal of Fungi. 2025; 11(5):346. https://doi.org/10.3390/jof11050346
Chicago/Turabian StyleBai, Ruxue, Qingchun Wang, and Haiying Bao. 2025. "Whole-Genome Characterization of Inonotus hispidus from Ulmus macrocarpa and Its Comparative Genomics with Strains from Morus alba and Acer truncatum" Journal of Fungi 11, no. 5: 346. https://doi.org/10.3390/jof11050346
APA StyleBai, R., Wang, Q., & Bao, H. (2025). Whole-Genome Characterization of Inonotus hispidus from Ulmus macrocarpa and Its Comparative Genomics with Strains from Morus alba and Acer truncatum. Journal of Fungi, 11(5), 346. https://doi.org/10.3390/jof11050346