

Harnessing Xylanase Potential in Thermothelomyces fergusii: Insights from Computational and Functional Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Screening of GH10, GH11, and GH43 Genes
2.2. Construction of Phylogenetic Tree of TfGH10, TfGH11, and TfGH43 Gene Families
2.3. Comprehensive Gene Structure Analysis of TfGH10, TfGH11, and TfGH43 Genes
2.4. Building of Protein 3D Structure and Protein–Ligand Interaction
2.5. Analysis of Gene Location, Duplication, Circos, and Gene Mutation
2.6. Analysis of Gene Enrichment and Hypothetical Model of Xylanase Enzyme Function
2.7. Fungal Strain Culturing Condition
2.8. Biochemical Analysis
2.9. RNA Extraction and Expression Analysis
3. Results
3.1. General Features of GH10, GH11, and GH43 Genes
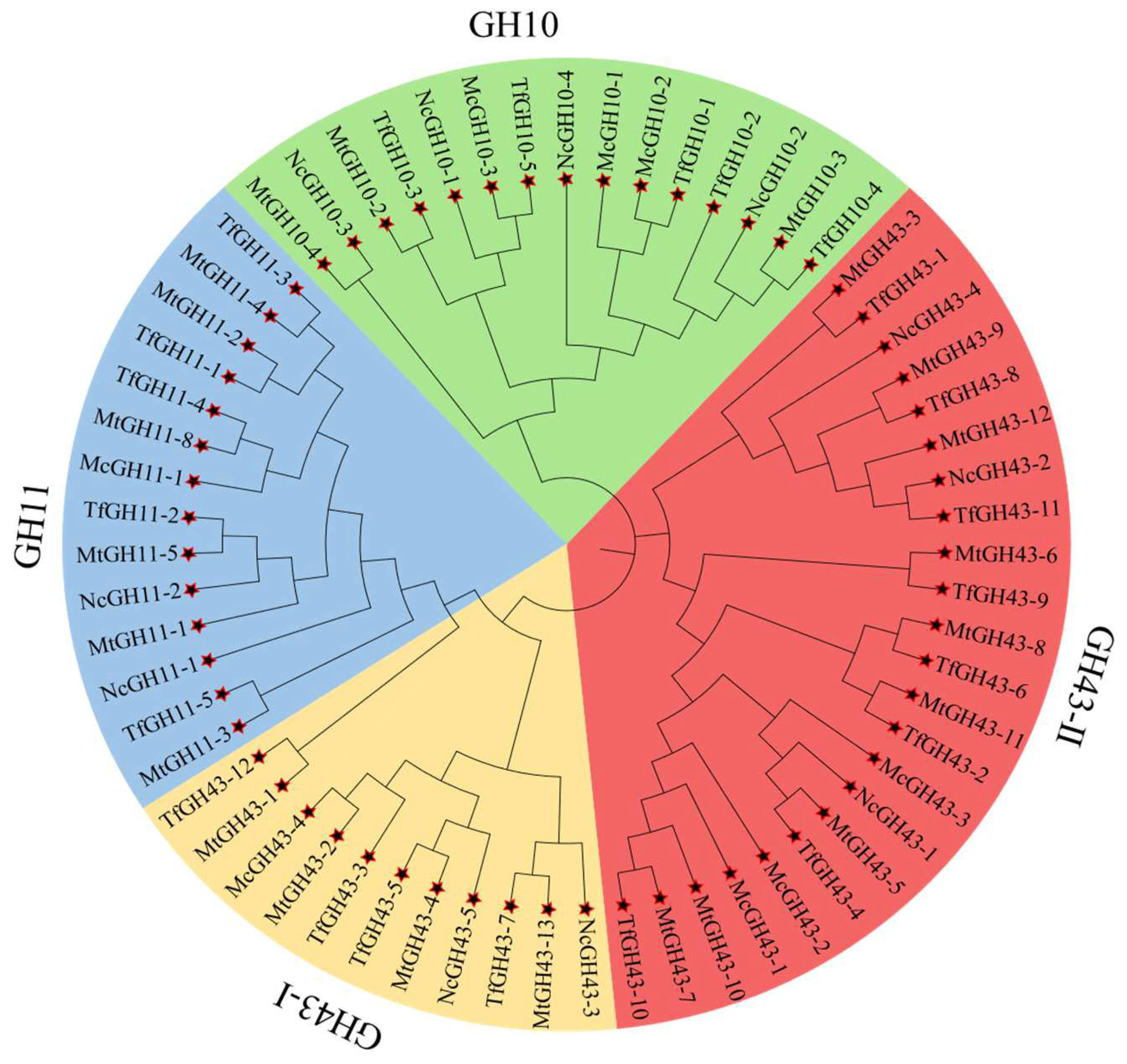
3.2. Phylogenetic Association of TfGH10, TfGH11, and TfGH43 Family Members
3.3. Gene Structure, Conserved Domain, and Motif Analysis of TfGH10, TfGH11, and TfGH43
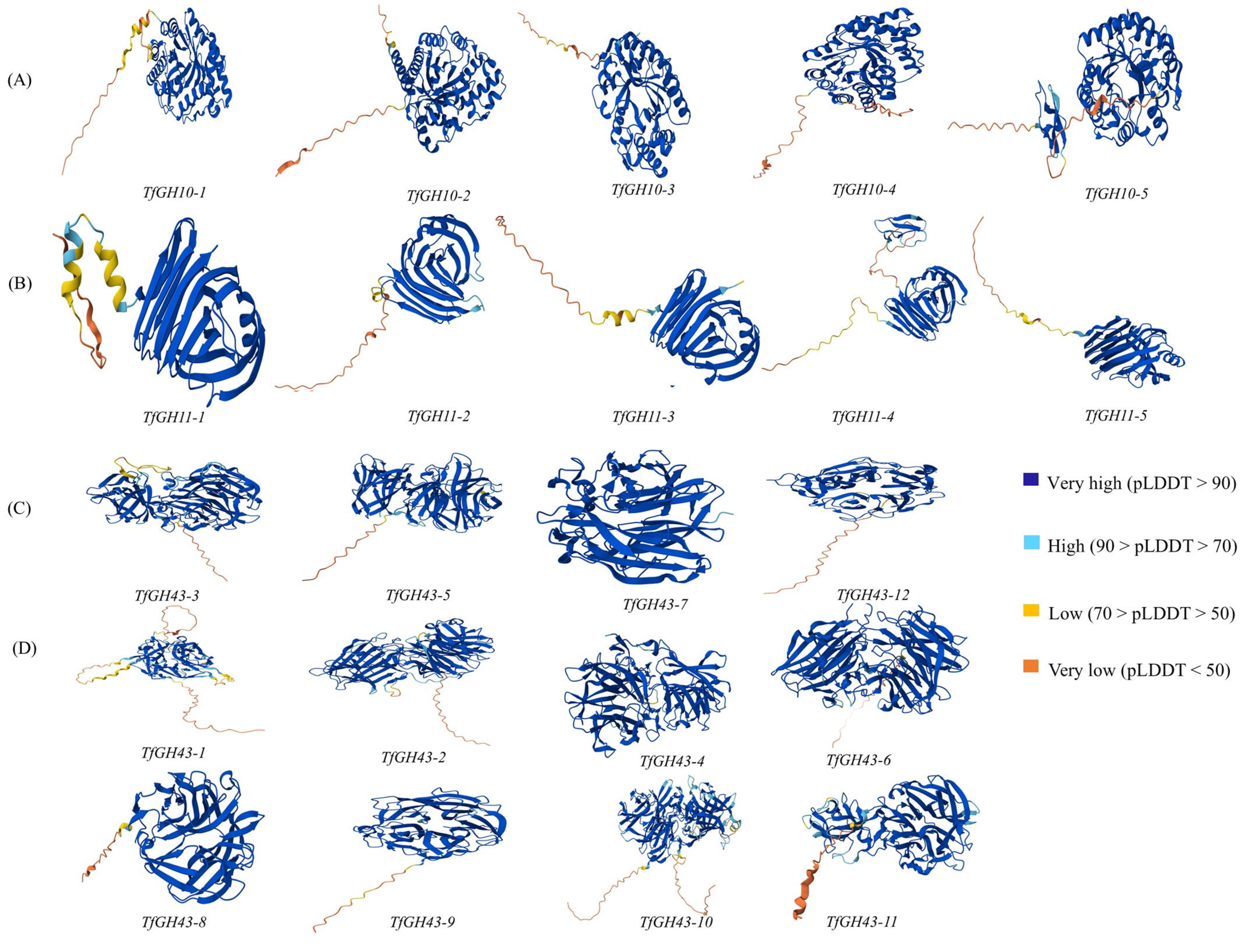
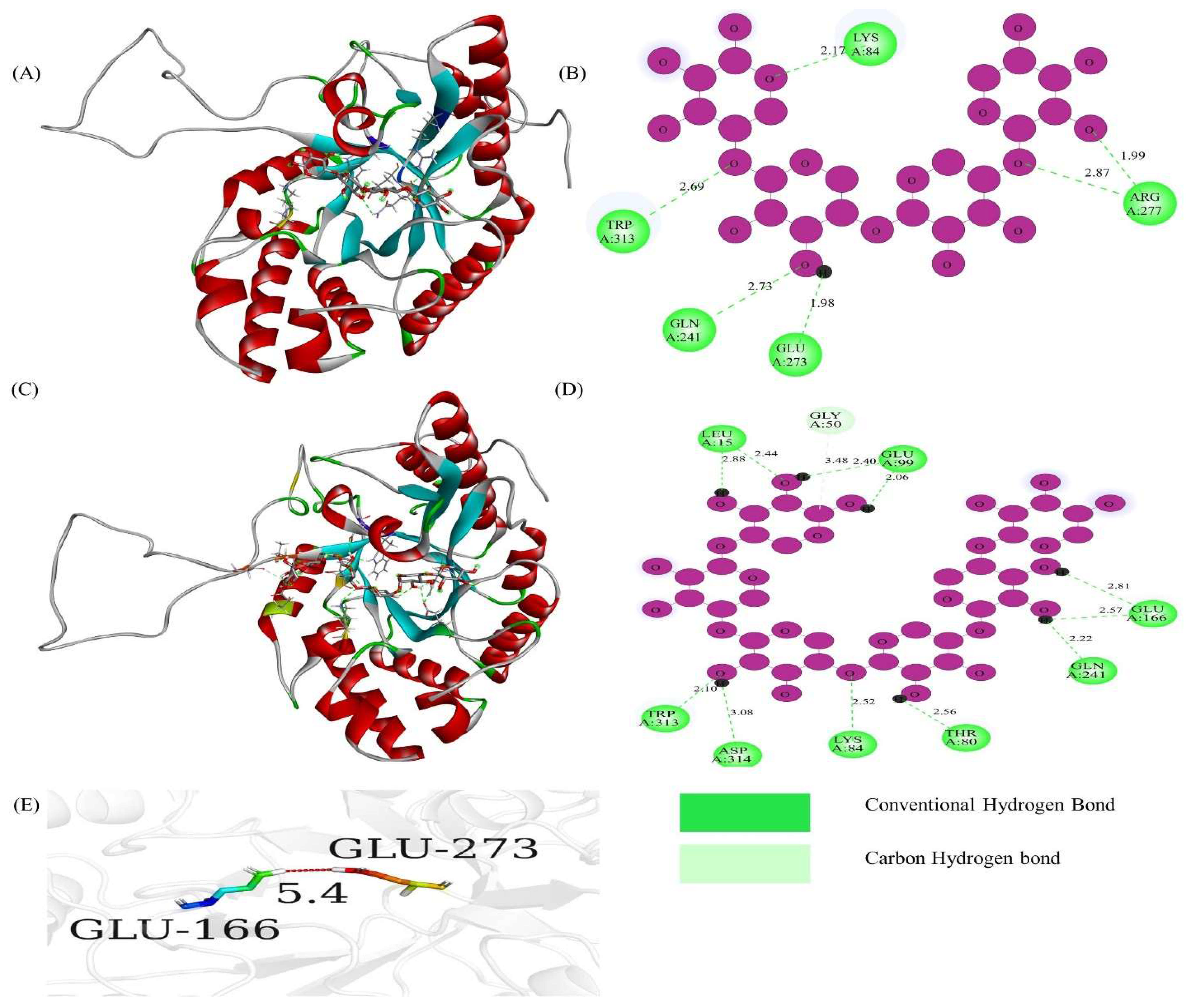
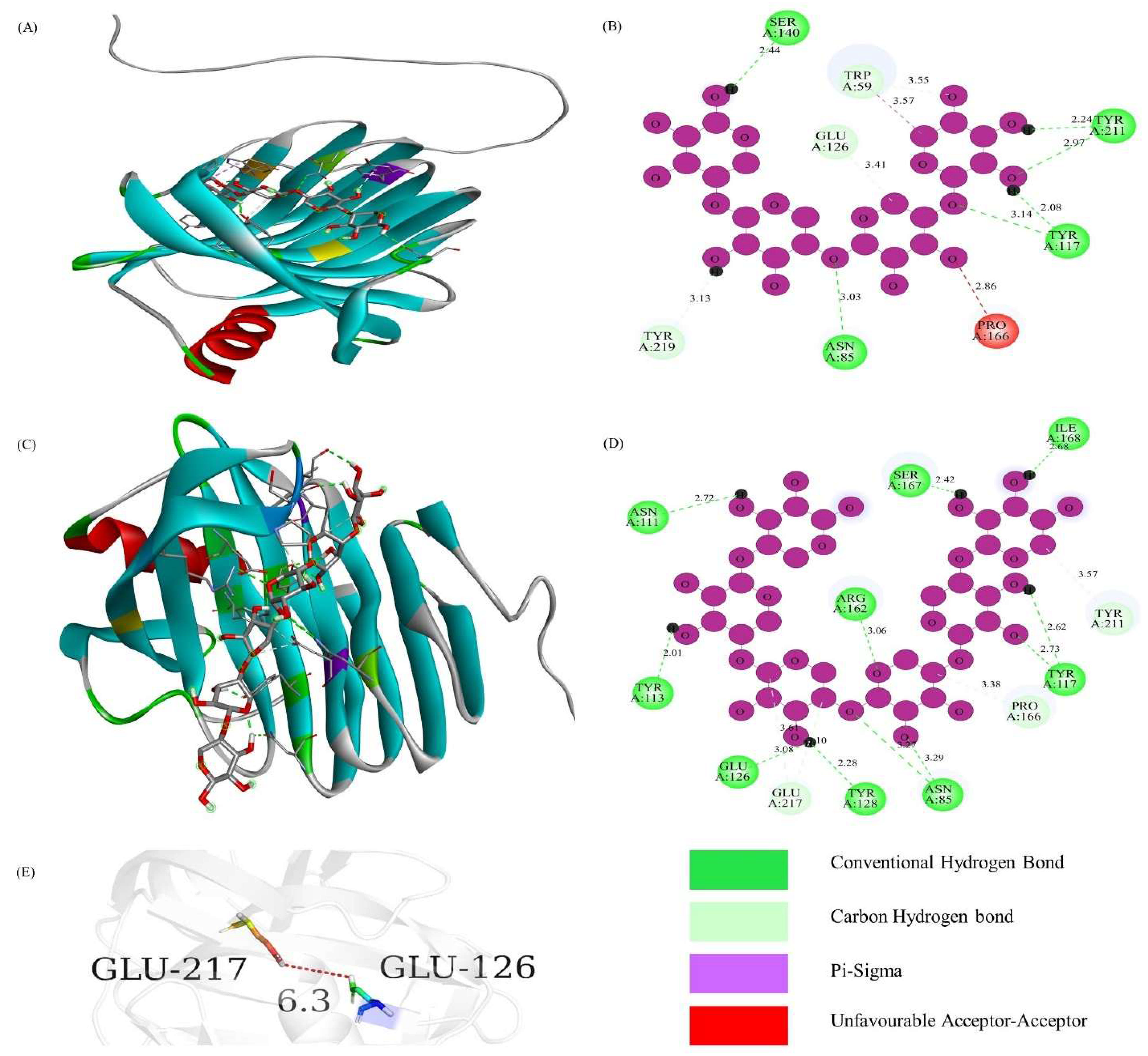
3.4. Prediction of Protein 3D Model and Catalytic Active Sites
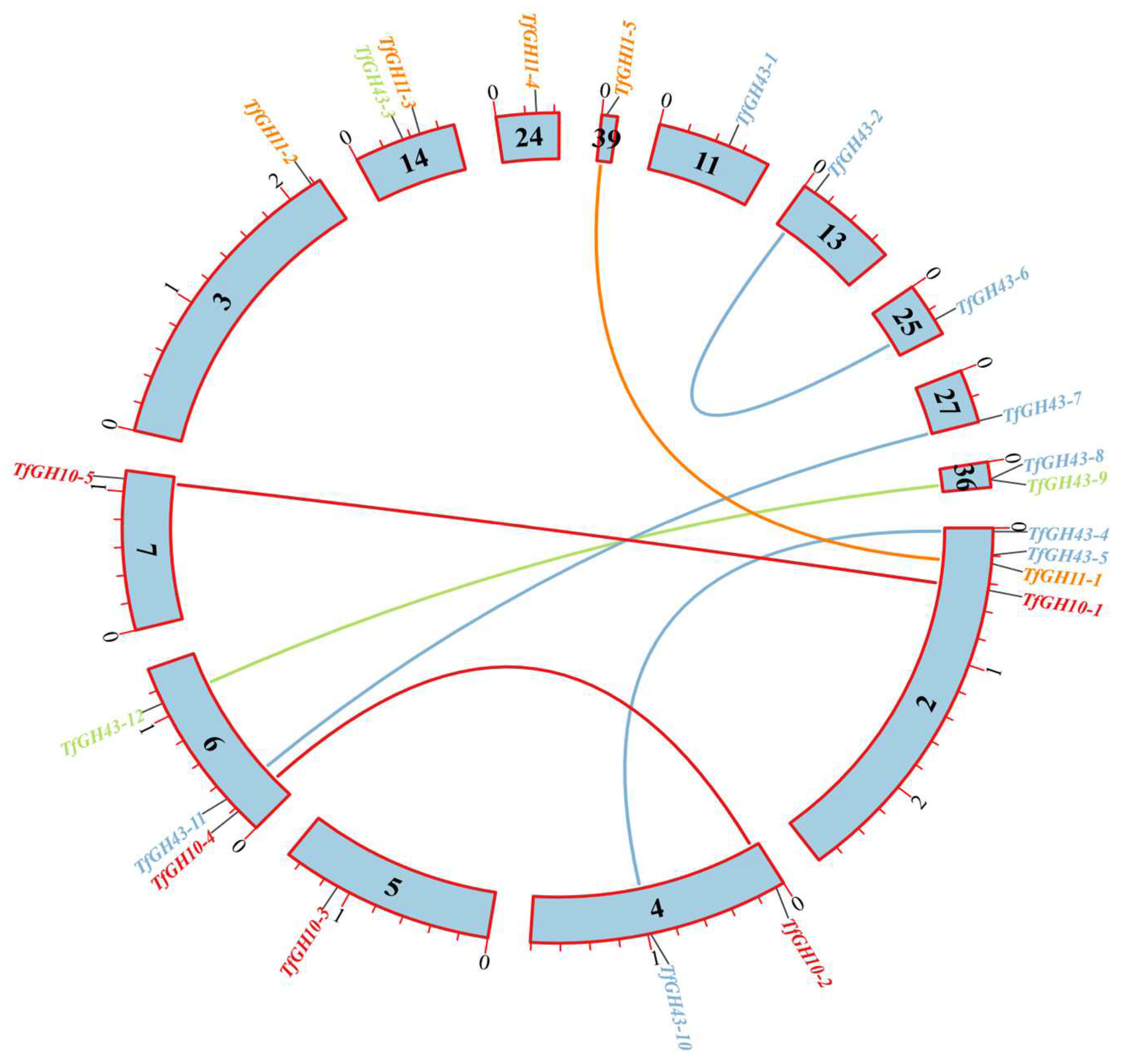
3.5. Gene Duplication, Scaffold Localization, and Ka/Ks Calculation
3.6. Xylanase GO Enrichment and Catalytic Reaction Models
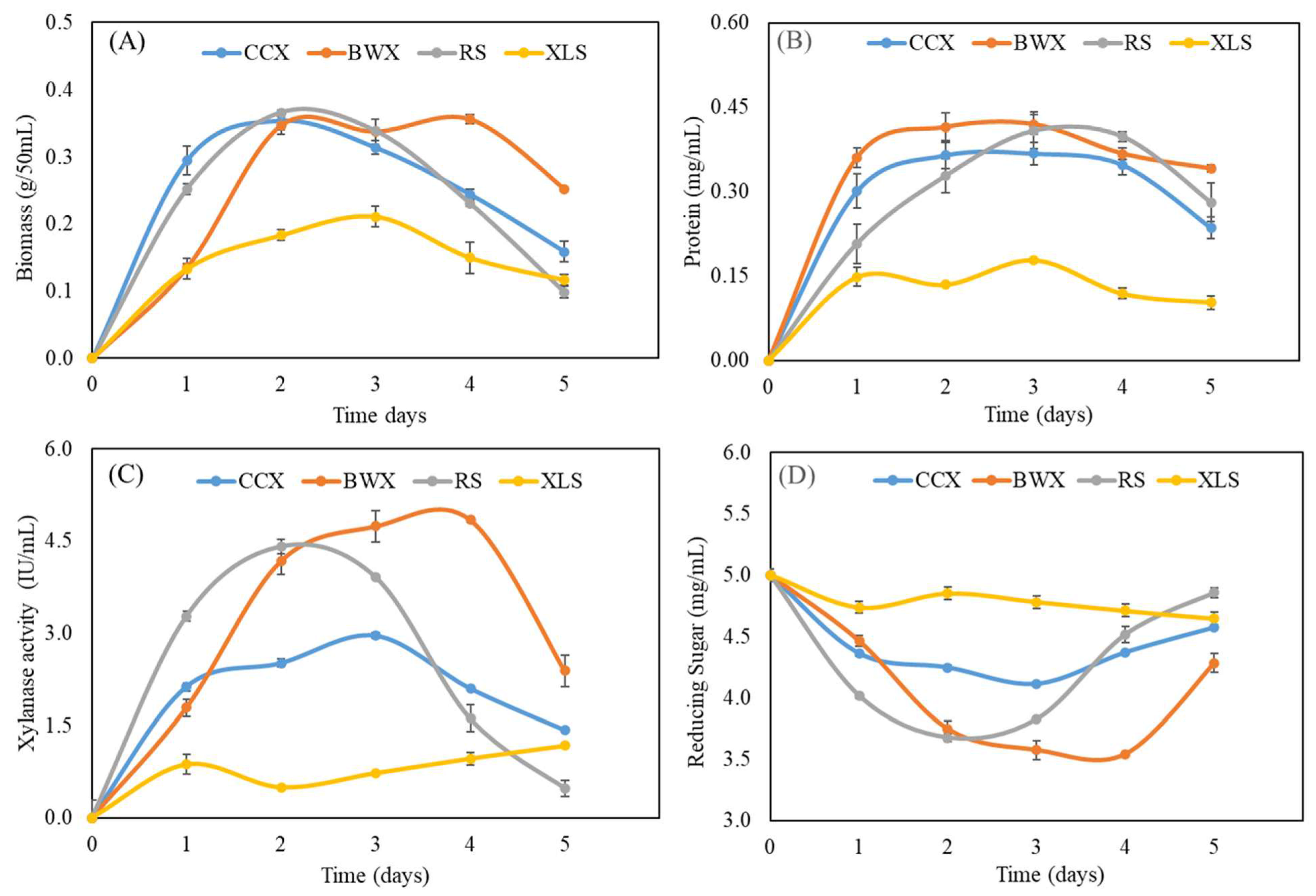
3.7. Analysis of Biochemical Properties in Response to Hemicellulose Sugar
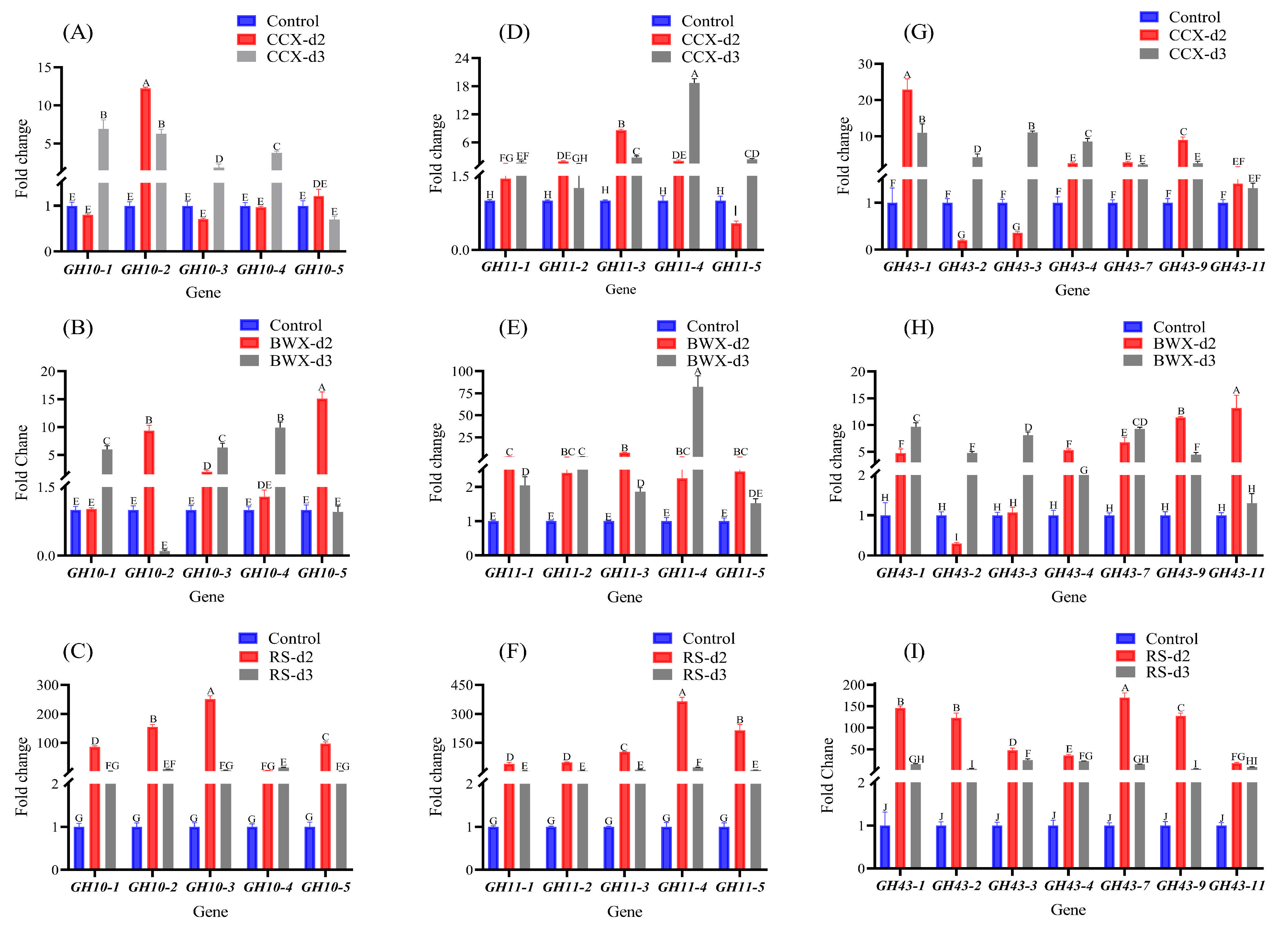
3.8. Gene Expression Analysis Under Hemicellulose Cultivation Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saha, B.C. Hemicellulose bioconversion. J. Ind. Microbiol. Biotechnol. 2003, 30, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.; Mohanty, P.; Pant, K.K.; Naik, S.; Kozinski, J.A.; Dalai, A.K. Characterization of North American lignocellulosic biomass and biochars in terms of their candidacy for alternate renewable fuels. BioEnergy Res. 2013, 6, 663–677. [Google Scholar] [CrossRef]
- Villas-Bôas, S.G.; Esposito, E.; Mitchell, D.A. Microbial conversion of lignocellulosic residues for production of animal feeds. Anim. Feed Sci. Technol. 2002, 98, 1–12. [Google Scholar] [CrossRef]
- Damm, T.; Commandeur, U.; Fischer, R.; Usadel, B.; Klose, H. Improving the utilization of lignocellulosic biomass by polysaccharide modification. Process Biochem. 2016, 51, 288–296. [Google Scholar] [CrossRef]
- Abedi, E.; Fatemi, F.; Sefidbakht, Y.; Siadat, S.E.R. Development and characterization of a thermostable GH11/GH10 xylan degrading chimeric enzyme. Enzym. Microb. Technol. 2021, 149, 109854. [Google Scholar] [CrossRef]
- Mendonça, M.; Barroca, M.; Collins, T. Endo-1, 4-β-xylanase-containing glycoside hydrolase families: Characteristics, singularities and similarities. Biotechnol. Adv. 2023, 65, 108148. [Google Scholar] [CrossRef]
- Chu, Y.; Hao, Z.; Wang, K.; Tu, T.; Huang, H.; Wang, Y.; Bai, Y.G.; Wang, Y.; Luo, H.; Yao, B.; et al. The GH10 and GH48 dual-functional catalytic domains from a multimodular glycoside hydrolase synergize in hydrolyzing both cellulose and xylan. Biotechnol. Biofuels 2019, 12, 279. [Google Scholar] [CrossRef]
- Paës, G.; Berrin, J.-G.; Beaugrand, J. GH11 xylanases: Structure/function/properties relationships and applications. Biotechnol. Adv. 2012, 30, 564–592. [Google Scholar] [CrossRef]
- Zhang, X.J.; Wang, L.; Wang, S.; Chen, Z.L.; Li, Y.H. Contributions and characteristics of two bifunctional GH43 β-xylosidase/α-L-arabinofuranosidases with different structures on the xylan degradation of Paenibacillus physcomitrellae strain XB. Microbiol. Res. 2021, 253, 126886. [Google Scholar] [CrossRef]
- Pang, S.L.; Wang, Y.Y.; Wang, L.; Zhang, X.J.; Li, Y.H. The CBM91 module enhances the activity of β-xylosidase/α-L-arabinofuranosidase PphXyl43B from Paenibacillus physcomitrellae XB by adopting a unique loop conformation at the top of the active pocket. Int. J. Biol. Macromol. 2024, 266, 131275. [Google Scholar] [CrossRef]
- Miao, Y.; Li, P.; Li, G.; Liu, D.; Druzhinina, I.S.; Kubicek, C.P.; Shen, Q.; Zhang, R. Two degradation strategies for overcoming the recalcitrance of natural lignocellulosic xylan by polysaccharides-binding GH 10 and GH 11 xylanases of filamentous fungi. Environ. Microbiol. 2017, 19, 1054–1064. [Google Scholar] [PubMed]
- Huang, Y.; Busk, P.K.; Lange, L. Cellulose and hemicellulose-degrading enzymes in Fusarium commune transcriptome and functional characterization of three identified xylanases. Enzym. Microb. Technol. 2015, 73, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Arioka, M. Functional analyses of xylanolytic enzymes involved in xylan degradation and utilization in Neurospora crassa. Int. J. Biol. Macromol. 2021, 169, 302–310. [Google Scholar] [CrossRef]
- Dodda, S.R.; Hossain, M.; Kapoor, B.S.; Dasgupta, S.; Aikat, K.; Mukhopadhyay, S.S. Computational approach for identification, characterization, three-dimensional structure modelling and machine learning-based thermostability prediction of xylanases from the genome of Aspergillus fumigatus. Comput. Biol. Chem. 2021, 91, 107451. [Google Scholar]
- Álvarez-Cervantes, J.; Díaz-Godínez, G.; Mercado-Flores, Y.; Gupta, V.K.; Anducho-Reyes, M.A. Phylogenetic analysis of β-xylanase SRXL1 of Sporisorium reilianum and its relationship with families (GH10 and GH11) of Ascomycetes and Basidiomycetes. Sci. Rep. 2016, 6, 24010. [Google Scholar]
- Morais, M.A.; Coines, J.; Domingues, M.N.; Pirolla, R.A.; Tonoli, C.C.; Santos, C.R.; Correa, J.B.; Gozzo, F.C.; Rovira, C.; Murakami, M.T. Two distinct catalytic pathways for GH43 xylanolytic enzymes unveiled by X-ray and QM/MM simulations. Nat. Commun. 2021, 12, 367. [Google Scholar]
- Basotra, N.; Joshi, S.; Satyanarayana, T.; Pati, P.K.; Tsang, A.; Chadha, B.S. Expression of catalytically efficient xylanases from thermophilic fungus Malbranchea cinnamomea for synergistically enhancing hydrolysis of lignocellulosics. Int. J. Biol. Macromol. 2018, 108, 185–192. [Google Scholar]
- Lopes, A.M.M.; Martins, M.; Goldbeck, R. Heterologous expression of lignocellulose-modifying enzymes in microorganisms: Current status. Mol. Biotechnol. 2021, 63, 184–199. [Google Scholar]
- Wang, H.; Li, Z.; Liu, H.; Li, S.; Qiu, H.; Li, K.; Luo, X.; Song, Y.; Wang, N.; He, H.; et al. Heterologous expression in Pichia pastoris and characterization of a novel GH11 xylanase from saline-alkali soil with excellent tolerance to high pH, high salt concentrations and ethanol. Protein Expr. Purif. 2017, 139, 71–77. [Google Scholar]
- Lu, Y.; Fang, C.; Wang, Q.; Zhou, Y.; Zhang, G.; Ma, Y. High-level expression of improved thermo-stable alkaline xylanase variant in Pichia Pastoris through codon optimization, multiple gene insertion and high-density fermentation. Sci. Rep. 2016, 6, 37869. [Google Scholar]
- Berka, R.M.; Grigoriev, I.V.; Otillar, R.; Salamov, A.; Grimwood, J.; Reid, I.; Ishmael, N.; John, T.; Darmond, C.; Moisan, M.-C.; et al. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat. Biotechnol. 2011, 29, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Hüttner, S.; Nguyen, T.T.; Granchi, Z.; Chin-A-Woeng, T.; Ahrén, D.; Larsbrink, J.; Thanh, V.N.; Olsson, L. Combined genome and transcriptome sequencing to investigate the plant cell wall degrading enzyme system in the thermophilic fungus Malbranchea cinnamomea. Biotechnol. Biofuels 2017, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Borin, G.P.; Sanchez, C.C.; de Santana, E.S.; Zanini, G.K.; Dos Santos, R.A.C.; de Oliveira Pontes, A.; de Souza, A.T.; Dal’Mas, R.M.M.T.S.; Riaño-Pachón, D.M.; Goldman, G.H.; et al. Comparative transcriptome analysis reveals different strategies for degradation of steam-exploded sugarcane bagasse by Aspergillus niger and Trichoderma reesei. BMC Genom. 2017, 18, 501. [Google Scholar] [CrossRef] [PubMed]
- Shallom, D.; Shoham, Y. Microbial hemicellulases. Curr. Opin. Microbiol. 2003, 6, 219–228. [Google Scholar] [CrossRef]
- Zhu, N.; Yang, J.; Ji, L.; Liu, J.; Yang, Y.; Yuan, H. Metagenomic and metaproteomic analyses of a corn stover-adapted microbial consortium EMSD5 reveal its taxonomic and enzymatic basis for degrading lignocellulose. Biotechnol. Biofuels 2016, 9, 243. [Google Scholar] [CrossRef]
- Morgenstern, I.; Powlowski, J.; Ishmael, N.; Darmond, C.; Marqueteau, S.; Moisan, M.-C.; Quenneville, G.; Tsang, A. A molecular phylogeny of thermophilic fungi. Fungal Biol. 2012, 116, 489–502. [Google Scholar] [CrossRef]
- Rosenberg, S. Cellulose and lignocellulose degradation by thermophilic and thermotolerant fungi. Mycologia 1978, 70, 1–13. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [PubMed]
- Studio, D. Discovery studio. Accelrys [2.1] 2008, 420, 1–9. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Chem. Biol. Methods Protoc. 2015, 1263, 243–250. [Google Scholar]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar]
- Chao, J.; Li, Z.; Sun, Y.; Aluko, O.O.; Wu, X.; Wang, Q.; Liu, G. MG2C: A user-friendly online tool for drawing genetic maps. Mol. Hortic. 2021, 1, 16. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar]
- Ashoor, S.; Sukumaran, R.K. Mild alkaline pretreatment can achieve high hydrolytic and fermentation efficiencies for rice straw conversion to bioethanol. Prep. Biochem. Biotechnol. 2020, 50, 814–819. [Google Scholar]
- Li, X.; Han, C.; Li, W.; Chen, G.; Wang, L. Insights into the cellulose degradation mechanism of the thermophilic fungus Chaetomium thermophilum based on integrated functional omics. Biotechnol. Biofuels 2020, 13, 143. [Google Scholar] [CrossRef]
- Božinović, M.; Sokač, T.; Šalić, A.; Dukarić, A.-M.; Tišma, M.; Planinić, M.; Zelić, B. Standardization of 3, 5-dinitrosalicylic acid (DNS) assay for measuring xylanase activity: Detecting and solving problems. Croat. J. Food Sci. Technol. 2023, 15, 151–162. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.; Liu, J.; Miao, T.; Zheng, F.; Rahim, K.; Lou, H.; Jiang, W. Characterization of two endo-β-1, 4-xylanases from Myceliophthora thermophila and their saccharification efficiencies, synergistic with commercial cellulase. Front. Microbiol. 2018, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, M.; Wei, S.; Wang, X.; Zhao, Y.; Han, Z.; Ye, X.; Li, Z.; Ji, Y.; Cui, Z.; et al. Characterization of a multi-domain exo-β-1, 3-galactanase from Paenibacillus xylanexedens. Int. J. Biol. Macromol. 2024, 266, 131413. [Google Scholar] [CrossRef]
- Ichinose, H.; Yoshida, M.; Fujimoto, Z.; Kaneko, S. Characterization of a modular enzyme of exo-1, 5-α-L-arabinofuranosidase and arabinan binding module from Streptomyces avermitilis NBRC14893. Appl. Microbiol. Biotechnol. 2008, 80, 399–408. [Google Scholar] [CrossRef]
- Kolbusz, M.A.; Di Falco, M.; Ishmael, N.; Marqueteau, S.; Moisan, M.-C.; da Silva Baptista, C.; Powlowski, J.; Tsang, A. Transcriptome and exoproteome analysis of utilization of plant-derived biomass by Myceliophthora thermophila. Fungal Genet. Biol. 2014, 72, 10–20. [Google Scholar] [CrossRef]
- Verma, D. Extremophilic prokaryotic endoxylanases: Diversity, applicability, and molecular insights. Front. Microbiol. 2021, 12, 728475. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, S.; Shang, W.; Yan, Z.; Wu, X.; Li, Y.; Chen, G.; Liu, X.; Wang, L. Synergistic mechanism of GH11 xylanases with different action modes from Aspergillus niger An76. Biotechnol. Biofuels 2021, 14, 118. [Google Scholar] [CrossRef]
- Yang, Q.; Gao, Y.; Huang, Y.; Xu, Q.; Luo, X.-M.; Liu, J.-L.; Feng, J.-X. Identification of three important amino acid residues of xylanase AfxynA from Aspergillus fumigatus for enzyme activity and formation of xylobiose as the major product. Process Biochem. 2015, 50, 571–581. [Google Scholar] [CrossRef]
- Qiu, J.; Han, H.; Sun, B.; Chen, L.; Yu, C.; Peng, R.; Yao, Q. Residue mutations of xylanase in Aspergillus kawachii alter its optimum pH. Microbiol. Res. 2016, 182, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stajich, J.E. Fungal genomes and insights into the evolution of the kingdom. Microbiol. Spectr. 2017, 5, 1128. [Google Scholar] [CrossRef]
- Miao, Y.; Liu, D.; Li, G.; Li, P.; Xu, Y.; Shen, Q.; Zhang, R. Genome-wide transcriptomic analysis of a superior biomass-degrading strain of A. fumigatus revealed active lignocellulose-degrading genes. BMC Genom. 2015, 16, 459. [Google Scholar]
- Sveholm, E.; Mattila, H.; Aro, N.; Valkonen, M.; Paasela, T.; Pakula, T.M. Transcriptomic and metabolic changes in Trichoderma reesei caused by mutation in xylanase regulator 1 (xyr1). Biotechnol. Biofuels Bioprod. 2024, 17, 106. [Google Scholar]
- Gupte, A.P.; Basaglia, M.; Casella, S.; Favaro, L. Rice waste streams as a promising source of biofuels: Feedstocks, biotechnologies and future perspectives. Renew. Sustain. Energy Rev. 2022, 167, 112673. [Google Scholar] [CrossRef]
- Basotra, N.; Kaur, B.; Di Falco, M.; Tsang, A.; Chadha, B.S. Mycothermus thermophilus (Syn. Scytalidium thermophilum): Repertoire of a diverse array of efficient cellulases and hemicellulases in the secretome revealed. Bioresour. Technol. 2016, 222, 413–421. [Google Scholar]
- Ribeiro, L.F.; De Lucas, R.C.; Vitcosque, G.L.; Ribeiro, L.F.; Ward, R.J.; Rubio, M.V.; Damásio, A.R.; Squina, F.M.; Gregory, R.C.; Walton, P.H. A novel thermostable xylanase GH10 from Malbranchea pulchella expressed in Aspergillus nidulans with potential applications in biotechnology. Biotechnol. Biofuels 2014, 7, 115. [Google Scholar]
- Gong, W.; Dai, L.; Zhang, H.; Zhang, L.; Wang, L. A highly efficient xylan-utilization system in Aspergillus niger An76: A functional-proteomics study. Front. Microbiol. 2018, 9, 430. [Google Scholar]
- Bala, A.; Anu; Alokika; Kumar, A.; Kumar, S.; Singh, D.; Singh, B. Secretome analysis of thermophilic mould Myceliophthora thermophila cultivated on rice straw and hydrolysis of lignocellulosic biomass for bioethanol production. Biocatal. Biotransf. 2020, 38, 283–292. [Google Scholar]
- Huang, Y.; Zheng, X.; Pilgaard, B.; Holck, J.; Muschiol, J.; Li, S.; Lange, L. Identification and characterization of GH11 xylanase and GH43 xylosidase from the chytridiomycetous fungus, Rhizophlyctis rosea. Appl. Microbiol. Biotechnol. 2019, 103, 777–791. [Google Scholar] [CrossRef]
- Tõlgo, M.; Hüttner, S.; Rugbjerg, P.; Thuy, N.T.; Thanh, V.N.; Larsbrink, J.; Olsson, L. Genomic and transcriptomic analysis of the thermophilic lignocellulose-degrading fungus Thielavia terrestris LPH172. Biotechnol. Biofuels 2021, 14, 131. [Google Scholar]
- Singhvi, M.S.; Gokhale, D.V. Lignocellulosic biomass: Hurdles and challenges in its valorization. Appl. Microbiol. Biotechnol. 2019, 103, 9305–9320. [Google Scholar] [PubMed]
- Gírio, F.M.; Fonseca, C.; Carvalheiro, F.; Duarte, L.C.; Marques, S.; Bogel-Łukasik, R. Hemicelluloses for fuel ethanol: A review. Bioresour. Technol. 2010, 101, 4775–4800. [Google Scholar] [PubMed]
- dos Santos Gomes, A.C.; Falkoski, D.; Battaglia, E.; Peng, M.; Nicolau de Almeida, M.; Coconi Linares, N.; Meijnen, J.-P.; Visser, J.; De Vries, R.P. Myceliophthora thermophila Xyr1 is predominantly involved in xylan degradation and xylose catabolism. Biotechnol. Biofuels 2019, 12, 220. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Gene ID | Gene ID Rename | Scaffold | Second Domain | EC Number | Genomic Position | SP | Gene Length | CDS (bp) | AA (bp) | E:I | SI% | SS% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_3198 | TfGH10-1 | scaffold_2 | - | 3.2.1.8 | 446,763–448,277− | 17 | 1515 | 1068 | 356 | 4:3 | 76.9 | 87.6 |
| gene_6450 | TfGH10-2 | scaffold_4 | - | 3.2.1.8 | 59,346–60,446+ | 19 | 1101 | 1101 | 367 | 1:0 | 78.6 | 85.4 |
| gene_7390 | TfGH10-3 | scaffold_5 | - | 3.2.1.8 | 1,092,416–1,093,212+ | 15 | 797 | 510 | 170 | 4:3 | 44.6 | 47.7 |
| gene_7581 | TfGH10-4 | scaffold_6 | - | 3.2.1.8 | 169,430–170,763+ | 18 | 1334 | 1122 | 374 | 3:2 | 87.2 | 93.6 |
| gene_8290 | TfGH10-5 | scaffold_7 | CBM1 | 3.2.1.8 | 1,092,564–1,093,928+ | 17 | 1365 | 1146 | 382 | 4:3 | 84.8 | 90.4 |
| gene_3142 | TfGH11-1 | scaffold_2 | - | 3.2.1.8 | 254,904–256,034+ | 16 | 1131 | 693 | 231 | 2:1 | 94.8 | 96.5 |
| gene_5897 | TfGH11-2 | scaffold_3 | - | 3.2.1.8 | 2,178,512–2,179,256+ | 18 | 745 | 666 | 222 | 2:1 | 81.9 | 91.9 |
| gene_2026 | TfGH11-3 | scaffold_14 | - | 3.2.1.8 | 467,894–468,688− | 20 | 795 | 687 | 229 | 2:1 | 88.2 | 93.4 |
| gene_4601 | TfGH11-4 | scaffold_24 | CBM1 | 3.2.1.8 | 277,861–278,860− | 19 | 1000 | 870 | 290 | 2:1 | 73 | 80.2 |
| gene_6409 | TfGH11-5 | scaffold_39 | - | 3.2.1.8 | 31,507–32,272− | 16 | 766 | 666 | 222 | 2:1 | 94.6 | 96.9 |
| gene_1275 | TfGH43-1 | scaffold_11 | - | - | 496,981–497,996+ | No | 1016 | 948 | 316 | 2:1 | 55.2 | 60 |
| gene_1647 | TfGH43-2 | scaffold_13 | CBM91 | 3.2.1.37 | 84,744–86,432− | 24 | 1689 | 1689 | 563 | 1:0 | 63.9 | 74.9 |
| gene_1989 | TfGH43-3 | scaffold_14 | CBM35 | - | 346,458–347,582+ | No | 1125 | 1125 | 375 | 1:0 | 74.5 | 75.1 |
| gene_3073 | TfGH43-4 | scaffold_2 | CBM91 | 3.2.1.37 | 25,833–27,446− | No | 1614 | 1614 | 538 | 1:0 | 97 | 99.1 |
| gene_3126 | TfGH43-5 | scaffold_2 | CBM35 | - | 193,250–195,023− | 18 | 1774 | 1356 | 452 | 5:4 | 90.7 | 97.8 |
| gene_4738 | TfGH43-6 | scaffold_25 | CBM91 | 3.2.1.37 | 282,021–283,700− | 23 | 1680 | 1680 | 560 | 1:0 | 85.9 | 91.1 |
| gene_5010 | TfGH43-7 | scaffold_27 | - | - | 379,672–380,655+ | No | 984 | 984 | 328 | 1:0 | 86.5 | 93 |
| gene_6310 | TfGH43-8 | scaffold_36 | - | - | 127,171–128,387− | 19 | 1217 | 963 | 321 | 4:3 | 93.1 | 96.9 |
| gene_6311 | TfGH43-9 | scaffold_36 | - | 3.2.1.99 | 129,332–130,498+ | 21 | 1167 | 963 | 321 | 3:2 | 90.3 | 95 |
| gene_6730 | TfGH43-10 | scaffold_4 | CBM91 | - | 980,624–982,408− | 23 | 1785 | 1785 | 595 | 1:0 | 81.9 | 85.8 |
| gene_7609 | TfGH43-11 | scaffold_6 | CBM42 | - | 286,430–287,869+ | 27 | 1440 | 1440 | 480 | 1:0 | 79.7 | 86.8 |
| gene_7865 | TfGH43-12 | scaffold_6 | - | - | 1,100,905–1,102,152+ | 25 | 1248 | 1065 | 355 | 4:3 | 91.5 | 95.5 |
| Gene Name | Xylotetraose (Kcal/mol) | Xylohexaose (Kcal/mol) |
|---|---|---|
| TfGH10-4 | −8.6 | −9.8 |
| TfGH11-1 | −9.5 | −9.9 |
| TfGH43-6 | −8.3 | −8.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waheed, A.; Chen, Y.; Su, Y.; Yan, Y.; Liu, G. Harnessing Xylanase Potential in Thermothelomyces fergusii: Insights from Computational and Functional Analysis. J. Fungi 2025, 11, 250. https://doi.org/10.3390/jof11040250
Waheed A, Chen Y, Su Y, Yan Y, Liu G. Harnessing Xylanase Potential in Thermothelomyces fergusii: Insights from Computational and Functional Analysis. Journal of Fungi. 2025; 11(4):250. https://doi.org/10.3390/jof11040250
Chicago/Turabian StyleWaheed, Abdul, Yi Chen, Ying Su, Yuxin Yan, and Gang Liu. 2025. "Harnessing Xylanase Potential in Thermothelomyces fergusii: Insights from Computational and Functional Analysis" Journal of Fungi 11, no. 4: 250. https://doi.org/10.3390/jof11040250
APA StyleWaheed, A., Chen, Y., Su, Y., Yan, Y., & Liu, G. (2025). Harnessing Xylanase Potential in Thermothelomyces fergusii: Insights from Computational and Functional Analysis. Journal of Fungi, 11(4), 250. https://doi.org/10.3390/jof11040250