
The Dual Pathogen Fusarium: Diseases, Incidence, Azole Resistance, and Biofilms




Abstract

1. Introduction
2. Clinical Types and Incidence of Human Fusariosis
2.1. Keratitis
2.2. Onychomycosis
2.3. Invasive Fusariosis (IF)
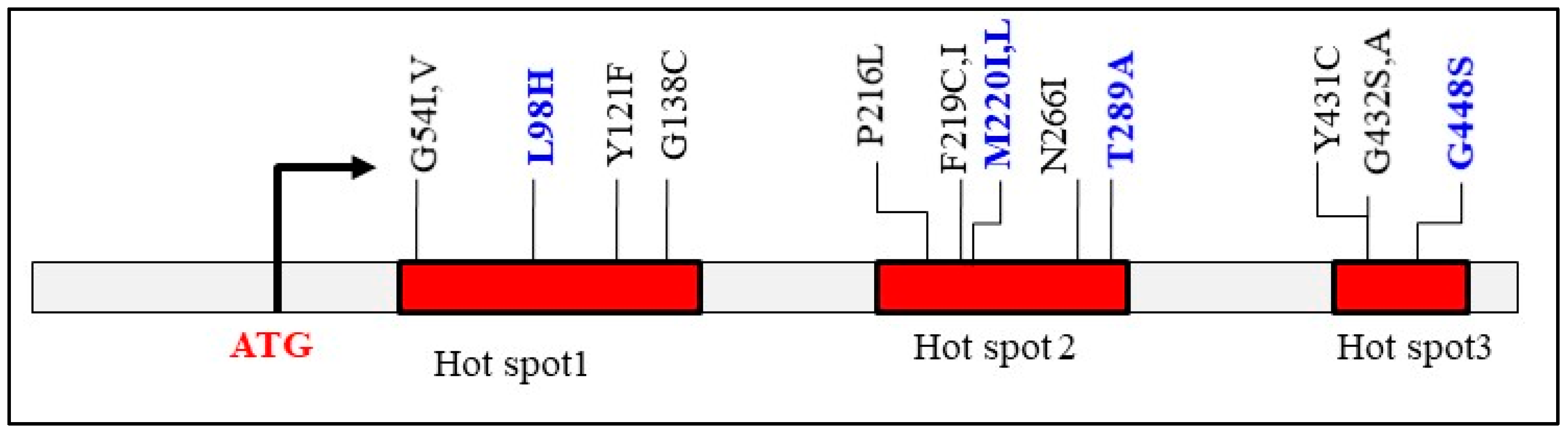
3. Intrinsic Azole Resistance of Fusarium
3.1. Resistance Evolution
3.2. Molecular Basis of Fusarium Azole Resistance
3.2.1. Point Mutations in CYP51 Paralogs
3.2.2. Overexpression of CYP51 Genes and Exclusion Drug by Efflux
3.2.3. Promoter Mutations in CYP51A and Their Potential Impact on Signal Transduction Pathways
4. Fusarium Biofilms
4.1. Biofilm Formation in Fusarium Infections in Humans
4.2. In Vivo and Ex Vivo Models of Fusarium Biofilm Formation
4.3. In Vitro Studies on Fusarium Biofilms
4.4. Biofilm Composition Analysis and Treatment Implications
5. Summary and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Diepeningen, A.D.; de Hoog, G.S. Challenges in Fusarium, a Trans-Kingdom Pathogen. Mycopathologia 2016, 181, 161–163. [Google Scholar] [CrossRef]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Casalini, G.; Giacomelli, A.; Antinori, S. The WHO fungal priority pathogens list: A crucial reappraisal to review the prioritisation. Lancet Microbe 2024, 7, 717–724. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- O’Donnell, K.; Sutton, D.A.; Rinaldi, M.G.; Magnon, K.C.; Cox, P.A.; Revankar, S.G.; Sanche, S.; Geiser, D.M.; Juba, J.H.; van Burik, J.A.; et al. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: Evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J. Clin. Microbiol. 2004, 42, 5109–5120. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, A. Current status of Fusarium and their management strategies. In Fusarium—An Overview on Current Status of the Genus; Mirmajlessi, S.M., Ed.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Chowdhary, C.; Kathuria, S.; Xu, J.; Meis, J.F. Emergence of azole resistant Aspergillus fumigatus strains due to agriculture azole use creates an increasing threat to human health. PLoS Pathog. 2013, 9, e1003633. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Invasive fusariosis. Clin. Microbiol. Rev. 2023, 36, e0015922. [Google Scholar] [CrossRef]
- James, J.E.; Lamping, E.; Santhanam, J.; Milne, T.J.; Abd Razak, M.F.; Zakaria, L.; Cannon, R.D. A 23 bp cyp51A promoter deletion associated with voriconazole resistance in clinical and environmental isolates of Neocosmospora keratoplastica. Front. Microbiol. 2020, 11, 272. [Google Scholar] [CrossRef]
- O’Donnell, K.; Ward, T.J.; Robert, V.A.R.G.; Crus, P.W.; Geisen, P.M.; Kang, S. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica 2015, 43, 583–595. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Guarner, J. Emerging and reemerging fungal infections. Semin. Diagn. Pathol. 2019, 36, 177–181. [Google Scholar] [CrossRef]
- Nucci, M.; Marr, K.A.; Vehreschild, M.J.; de Souza, C.A.; Velasco, E.; Cappellano, P.; Carlesse, F.; Queiroz-Telles, F.; Sheppard, D.C.; Kindo, A.; et al. Improvement in the outcome of invasive fusariosis in the last decade. Clin. Microbiol. Infect. 2014, 20, 580–585. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Cutaneous infection by Fusarium species in healthy and immunocompromised hosts: Implications for diagnosis and management. Clin. Infect. Dis. 2002, 35, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Nucci, M.; Anaissie, E. Fusarium infections in immunocompromised patients. Clin. Microbiol. Rev. 2007, 20, 695–704. [Google Scholar] [CrossRef]
- Hoffman, J.J.; Burton, M.J.; Leck, A. Mycotic keratitis-A global threat from the filamentous Fungi. J. Fungi 2021, 7, 273. [Google Scholar] [CrossRef] [PubMed]
- Oechsler, R.A.; Fellmeter, M.R.; Miller, D. Fusarium keratitis genotyping, in vitro susceptibility, and clinical outcomes. Cornea 2013, 32, 667–673. [Google Scholar] [CrossRef]
- Ranawaka, R.R.; Nagahawatte, A.; Gunasekara, T.A. Fusarium onychomycosis: Prevalence, clinical presentations, response to itraconazole and terbinafine pulse therapy, and 1-year follow-up in nine cases. Int. J. Dermatol. 2015, 54, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.L.; Knoeri, M.J.; Bourcier, T.; Merabet, L.; Borderie, V.M. Infection keratitis following corneal transplantation: A long-term cohort study. Clin. Exp. Ophthalmol. 2024, 52, 402–415. [Google Scholar] [CrossRef]
- Thomas, B.; Audonneau, N.C.; Machouart, M.; Debourgogne, A. Fusarium infections: Epidemiology aspects over 10 yrs in a university hospital in France. J. Infect. Public Health 2020, 13, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Kamwiziku, G.; Oladele, R.O.; Burton, M.J.; Prajna, N.V.; Leitman, T.M.; Denning, D.W. The case for fungal keratitis to be accepted as a neglected tropical disease. J. Fungi 2022, 8, 1047. [Google Scholar] [CrossRef]
- Green, M.; Apel, A.; Stapleton, F. Risk factors and causative organisms in microbial keratitis. Cornea 2008, 27, 22–27. [Google Scholar] [CrossRef]
- Brown, L.; Leck, A.K.; Gichangi, M.; Burton, M.J.; David, D.W. The global incidence and diagnosis of fungal keratitis. Lancet Infect. Dis. 2021, 21, e49–e57. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Z.; Cao, K.; Zhang, Z.; Xu, X.; Liang, Q. Trends of ocular fungal infections in North China (2001–2020). J. Infect. Public Health 2023, 16, 71–77. [Google Scholar] [CrossRef]
- Chang, D.C.; Grant, G.B.; O’Donnell, K.; Wannemuehler, K.A.; Noble-Wang, J.; Rao, C.Y.; Jacobson, L.M.; Crowell, C.S.; Sneed, R.S.; Lewis, M.F.T.; et al. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA 2006, 296, 953–963. [Google Scholar] [CrossRef]
- Szaliński, M.; Zgryźniak, A.; Rubisz, I.; Gajdzis, M.; Kaczmarek, R.; Przeździecka-Dołyk, J. Fusarium keratitis—review of current treatment possibilities. J. Clin. Med. 2021, 10, 5468. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.F.; Jin, X.Y.; Wang, X.L.; Sun, X.G. Effect of topical application of terbinafine on fungal keratitis. Chin. Med. J. 2009, 122, 1884–1888. [Google Scholar] [PubMed]
- Feng, Y.; Yang, Z.; Li, D.; Li, J.; Li, D.; de Hoog, S.; Shi, D. Nails and skin co-infection by Fusarium verticillioides and Proteus vulgaris secondary to arterial occlusion of lower extremity. Rev. Iberoam. Micol. 2024, 41, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Taborda, V.B.A.; Taborda, P.R.O.; Shemer, A.; Summerbell, R.C.; Nakrieko, K.A. High prevalence of mixed infections in global onychomycosis. PLoS ONE 2020, 15, e0239648. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wang, T.; Cooper, E.A.; Lincoln, S.A.; Foreman, H.C.; Scherer, W.P.; Bakotic, W.L. Clinical diagnosis and laboratory testing of abnormal appearing toenails: A retrospective assessment of confirmatory testing for onychomycosis in the United States, 2022–2023. J. Fungi 2024, 10, 149. [Google Scholar] [CrossRef]
- Mima, Y.; Yamamoto, M.; Makimura, K.; Iozumi, K. Onychomycosis with gray-green staining caused by Fusarium solani. Med. Mycol. Case Rep. 2024, 46, 100684. [Google Scholar] [CrossRef]
- Uemura, E.V.G.; Barbosa, M.D.S.; Simionatto, S.; Al-Harrasi, A.; Al-Hatmi, A.M.S.; Rossato, L. Onychomycosis caused by Fusarium species. J. Fungi 2022, 8, 360. [Google Scholar] [CrossRef]
- Tikvesli, M.; Gürcan, S.; Afyoncu, E.; Ürün, Y.G.; Solak, S.S. Mixed onychomycosis case with Fusarium solani: A case report. J. Immunol. Clin. Microbiol. 2022, 7, 45–50. [Google Scholar]
- Gupta, A.K.; Daigle, D.; Carviel, J.L. Evidence for biofilms in onychomycosis. J. Am. Acad. Dermatol. 2016, 74, 1241–1246. [Google Scholar] [CrossRef]
- Veiga, F.F.; de Castro-Hoshino, L.V.; Sato, F.; Baesso, M.L.; Silva, S.; Negri, M.; Svidzinski, T.I.E. Characterization of a biofilm formed by Fusarium oxysporum on the human nails. Int. J. Dermatol. 2022, 61, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, K.; Di Pietro, A.; Gow, N.A.; MacCallum, D. Murine model for Fusarium oxysporum invasive fusariosis reveals organ-specific structures for dissemination and long-term persistence. PLoS ONE 2014, 9, e89920. [Google Scholar] [CrossRef]
- Girmenia, C.; Pagano, L.; Corvatta, L.; Mele, L.; del Favero, A.; Martino, P. The epidemiology of fusariosis in patients with haematological diseases. Gimema Infection Programme. Br. J. Haematol. 2000, 111, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.P.; Marr, K.A.; Park, B.J.; Alexander, B.D.; Anaissie, E.J.; Walsh, T.J.; Ito, J.; Andes, D.R.; Baddley, J.W.; Brown, J.M.; et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: Overview of the transplant-associated infection surveillance network (transnet) database. Clin. Infect. Dis. 2010, 50, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Atalla, A.; Garnica, M.; Maiolino, A.; Nucci, M. Risk factors for invasive mold diseases in allogeneic hematopoietic cell transplant recipients. Transpl. Infect. Dis. 2015, 17, 7–13. [Google Scholar] [CrossRef]
- Nucci, M.; Varon, A.G.; Garnica, M.; Akiti, T.; Barreiros, G.; Trope, B.; Nouér, S.A. increased incidence of invasive fusariosis with cutaneous portal of entry. Braz. Emerg. Infect. Dis. 2013, 19, 1567–1572. [Google Scholar] [CrossRef]
- Rosa, P.D.D.; Ramirez-Castrillon, M.; Borges, R.; Aquino, V.; Fuentefria, M.A.; Goldani, Z.L. Epidemiological aspects and characterization of the resistance profile of Fusarium spp. in patients with invasive fusariosis. J. Med. Microbiol. 2019, 68, 1489–1496. [Google Scholar] [CrossRef]
- Pérez-Nadales, E.; Alastruey-Izquierdo, A.; Linares-Sicilia, M.J.; Soto-Debrán, J.C.; Abdala, E.; García-Rodríguez, J.; Montejo, M.; Muñoz, P.; Lletí, M.S.; Rezusta, A.; et al. Spanish fusariosis study group. invasive fusariosis in nonneutropenic patients, Spain, 2000–2015. Emerg. Infect. Dis. 2021, 27, 26–35. [Google Scholar] [CrossRef]
- Demonchy, J.; Biard, L.; Clere-Jehl, R.; Wallet, F.; Mokart, D.; Moreau, A.S.; Argaud, L.; Verlhac, C.; Pène, F.; Lautrette, A.; et al. Multicenter retrospective study of invasive fusariosis in intensive care units, France. Emerg. Infect. Dis. 2024, 30, 215–224. [Google Scholar] [CrossRef]
- Rychert, J. Benefits and limitations of maldi-tof mass spectrometry for the identification of microorganisms. J. Infect. 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Hachem, R.Y.; Kontoyiannis, D.P.; Chemaly, R.F.; Jiang, Y.; Reitzel, R.; Raad, I. Utility of galactomannan enzyme immunoassay and (1,3) beta-D-glucan in diagnosis of invasive fungal infections: Low sensitivity for Aspergillus fumigatus infection in hematologic malignancy patients. J. Clin. Microbiol. 2009, 47, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Dellière, S.; Guitard, J.; Marcela Sabou, M.; Angebault, C.; Moniot, M.; Cornu, M.; Hamane, S.; Bougnoux, M.E.; Imbert, S.; Pasquier, G.; et al. Detection of circulating DNA for the diagnosis of invasive fusariosis: Retrospective analysis of 15 proven cases. Med. Mycol. 2022, 60, myac049. [Google Scholar] [CrossRef] [PubMed]
- Pintye, A.; Bacsó, R.; Kovács, G.M. Trans-kingdom fungal pathogens infecting both plants and humans, and the problem of azole fungicide resistance. Front. Microbiol. 2024, 15, 1354757. [Google Scholar] [CrossRef]
- Bastos, R.W.; Rossato, L.; Goldman, G.H.; Santos, D.A. Fungicide effects on human fungal pathogens: Cross-resistance to medical drugs and beyond. PLoS Pathog. 2021, 17, e1010073. [Google Scholar] [CrossRef]
- Toda, M.; Beer, K.D.; Kuivila, K.M.; Chiller, T.M.; Jackson, B.R. Trends in agricultural triazole fungicide use in the united states, 1992-2016 and possible implications for antifungal-resistant fungi in human disease. Environ. Health Perspect. 2021, 129, 55001. [Google Scholar] [CrossRef]
- Kleinkauf, N.; Verweij, P.E.; Arendrup, M.C.; Donnelly, P.J.; Cuenca-Estrella, M.; Fraaije, B.; Melchers, W.J.G.; Adriaenssens, N.; Kema, G.H.J.; Ullmann, A.; et al. Risk Assessment on the Impact of Environmental Usage of Triazoles on the Development and Spread of Resistance to Medica Triazoles in Aspergillus Species; European Center for Disease and Control: Stockholm, Sweden, 2013. [Google Scholar] [CrossRef]
- Jørgensen, L.N.; Heick, T.M. Azole use in agriculture, horticulture, and wood preservation—Is it indispensable? Front. Cell Infect. Microbiol. 2021, 11, 730297. [Google Scholar] [CrossRef]
- Bilska, K.; Jurczak, S.; Kulik, T.; Ropelewska, E.; Olszewski, J.; Żelechowski, M.; Zapotoczny, P. Species composition and trichothecene genotype profiling of Fusarium field isolates recovered from wheat in Poland. Toxins 2018, 10, 325. [Google Scholar] [CrossRef]
- Ma, L.J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef]
- Srinivas, C.; Nirmala, D.D.; Narasimha, M.K.; Mohan, C.D.; Lakshmeesha, T.R.; Singh, B.; Kalagatur, N.K.; Niranjana, S.R.; Hashem, A.; Alqarawi, A.A.; et al. Fusarium oxysporum f. sp. lycopersici causal agent of vascular wilt disease of tomato: Biology to diversity—A review. Saudi J. Biol. Sci. 2019, 26, 1315–1324. [Google Scholar] [CrossRef]
- Pegg, K.G.; Coates, L.M.; O’Neill, W.T.; Turner, D.W. The epidemiology of Fusarium wilt of banana. Front. Plant Sci. 2019, 10, 1395. [Google Scholar] [CrossRef]
- Assress, H.A.; Selvarajan, R.; Nyoni, H.; Ogola, H.J.O.; Mamba, B.B.; Msagati, T.A.M. Azole antifungal resistance in fungal isolates from wastewater treatment plant effluents. Environ. Sci. Pollut. Res. Int. 2021, 28, 3217–3229. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, P.; Gruez, A.; Babin, A.L.; Frippiat, J.P.; Machouart, M.; Debourgogne, A. CYP51 Mutations in the Fusarium solani species complex: First clue to understand the low susceptibility to azoles of the genus Fusarium. J. Fungi 2022, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Al-Hatmi, A.M.; Meis, J.F.; Sybren de Hoog, G. Fusarium: Molecular diversity and intrinsic drug resistance. PLoS Pathog. 2016, 12, e1005464. [Google Scholar] [CrossRef]
- Al-Hatmi, A.M.S.; Bonifaz, A.; Ranque, S.; Sybren de Hoog, G.; Verweij, P.E.; Meis, J.F. Current antifungal treatment of fusariosis. Int. J. Antimicrob. Agents 2018, 51, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Dudakova, A.; Spiess, B.; Tangwattanachuleeporn, M.; Sasse, C.; Buchheidt, D.; Weig, M.; Groß, U.; Bader, O. Molecular tools for the detection and deduction of azole antifungal drug resistance phenotypes in Aspergillus species. Clin. Microbiol. Rev. 2017, 30, 1065–1091. [Google Scholar] [CrossRef]
- Zheng, B.; Yan, L.; Liang, W.; Yang, Q. Paralogous Cyp51s mediate the differential sensitivity of Fusarium oxysporum to sterol demethylation inhibitors. Pest. Manag. Sci. 2019, 75, 396–404. [Google Scholar] [CrossRef]
- Bader, O.; Weig, M.; Reichard, U.; Lugert, R.; Kuhns, M.; Christner, M.; Held, J.; Peter, S.; Schumacher, U.; Buchheidt, D.; et al. Cyp51A-based mechanisms of Aspergillus fumigatus azole drug resistance present in clinical samples from Germany. Antimicrob. Agents Chemother. 2013, 57, 3513–3517. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Gil, V.G.; Gutierrez, F.; Lindner, J.R.; Albataineh, M.T.; McCarthy, D.I.; Sanders, C.; Fan, H.; Fothergill, A.W.; Sutton, D.A. First detection of TR34 L98H and TR46 Y121F T289A Cyp51 mutations in Aspergillus fumigatus isolates in the united states. J. Clin. Microbiol. 2016, 54, 168–171. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, X.; Li, B.; Ma, Z. Characterization of sterol demethylation inhibitor-resistant isolates of Fusarium asiaticum and F. graminearum collected from wheat in China. Phytopathology 2009, 99, 487–497. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, J.; Guo, X.; Qian, L.; Xu, J.; Che, Z.; Chen, G.; Liu, S. Sensitivity and resistance risk assessment of Fusarium graminearum from wheat to prothioconazole. Plant Dis. 2022, 106, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, M.; Zhao, H.; Lu, F.; Wang, J.; Zhou, M. Molecular and biological characteristics of laboratory metconazole-resistant mutants in Fusarium graminearum. Pestic. Biochem. Physiol. 2018, 152, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Arikan, S.; Lozano-Chiu, M.; Paetznick, V.; Nangia, S.; Rex, J.H. Microdilution susceptibility testing of Amphotericin B, Itraconazole, and Voriconazole against clinical isolates of Aspergillus and Fusarium species. J. Clin. Microbiol. 1999, 37, 3946–3951. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Chang, Y.C. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 2012, 8, e1003022. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Sun, L.L.; Fu, B.Q.; Deng, J.; Jia, C.L.; Miao, M.X.; Yang, F.; Cao, Y.B.; Yan, T.H. Aneuploidy underlies brefeldin A-induced antifungal drug resistance in Cryptococcus neoformans. Front. Cell Infect. Microbiol. 2024, 14, 1397724. [Google Scholar] [CrossRef]
- Chang, Z.; Billmyre, R.B.; Lee, S.C.; Heitman, J. Broad antifungal resistance mediated by RNAi-dependent epimutation in the basal human fungal pathogen Mucor circinelloides. PLoS Genet. 2019, 15, e1007957. [Google Scholar] [CrossRef]
- Calo, S.; Nicolás, F.E.; Lee, S.C.; Vila, A.; Cervantes, M.; Torres-Martinez, S.; Ruiz-Vazquez, R.M.; Cardenas, M.E.; Heitman, J. A non-canonical RNA degradation pathway suppresses RNAi-dependent epimutations in the human fungal pathogen Mucor circinelloides. PLoS Genet. 2017, 13, e1006686. [Google Scholar] [CrossRef]
- Macedo, D.; Leonardelli, F.; Gamarra, S.; Garcia-Effron, G. Emergence of triazole resistance in Aspergillus spp. in Latin America. Curr. Fungal Infect. Rep. 2021, 15, 93–103. [Google Scholar] [CrossRef]
- Liu, M.; Zheng, N.; Li, D.; Zheng, H.; Zhang, L.; Ge, H.; Liu, W. Cyp51A-based mechanism of azole resistance in Aspergillus fumigatus: Illustration by a new 3D structural model of Aspergillus fumigatus CYP51A protein. Med. Mycol. 2016, 54, 400–408. [Google Scholar] [CrossRef]
- Gonzalez-Jimenez, I.; Lucio, J.; Amich, J.; Cuesta, I.; Sanchez Arroyo, R.; Alcazar-Fuoli, L.; Mellado, E. A Cyp51B mutation contributes to azole resistance in Aspergillus fumigatus. J. Fungi 2020, 6, 315. [Google Scholar] [CrossRef]
- Liu, W.; Sun, Y.; Chen, W.; Liu, W.; Wan, Z.; Bu, D.; Li, R. The T788G mutation in the cyp51C gene confers voriconazole resistance in Aspergillus flavus causing aspergillosis. Antimicrob. Agents Chemother. 2012, 56, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhou, X.; Jiao, Y.; Han, A.; Su, H.; Wang, L.; Zhou, H.; Li, W.; Liu, R. Potential mechanisms of hexaconazole resistance in Fusarium graminearum. Plant Dis. 2024, 108, 3133–3145. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, F.; Schnabel, G.; Wu, J.; Wang, Z.; Ma, Z. Paralogous cyp51 genes in Fusarium graminearum mediate differential sensitivity to sterol demethylation inhibitors. Fungal Genet. Biol. 2011, 48, 113–123. [Google Scholar] [CrossRef]
- Mair, W.J.; Deng, W.; Mullins, J.G.; West, S.; Wang, P.; Besharat, N.; Ellwood, S.R.; Oliver, R.P.; Lopez-Ruiz, F.J. Demethylase inhibitor fungicide resistance in Pyrenophora teres f. sp. teres associated with target site modification and inducible overexpression of Cyp51. Front. Microbiol. 2016, 7, 1279. [Google Scholar] [CrossRef]
- Jayawardana, M.A.; Fernando, W.G.D. the mechanisms of developing fungicide resistance in Fusarium graminearum causing Fusarium head blight and fungicide resistance management. Pathogens 2024, 13, 1012. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ortuño, D.; Loza-Reyes, E.; Atkins, S.L.; Fraaije, B.A. The CYP51C gene, a reliable marker to resolve interspecific phylogenetic relationships within the Fusarium species complex and a novel target for species-specific PCR. Int. J. Food Microbiol. 2010, 144, 301–309. [Google Scholar] [CrossRef]
- Abou Ammar, G.; Tryono, R.; Döll, K.; Karlovsky, P.; Deising, H.B.; Wirsel, S.G. Identification of ABC transporter genes of Fusarium graminearum with roles in azole tolerance and/or virulence. PLoS ONE 2013, 8, e79042. [Google Scholar] [CrossRef]
- Ma, T.; Li, Y.; Lou, Y.; Shi, J.; Sun, K.; Ma, Z.; Yan, L.; Yin, Y. The drug H+ antiporter FgQdr2 is essential for multiple drug resistance, ion homeostasis, and pathogenicity in Fusarium graminearum. J. Fungi 2022, 8, 1009. [Google Scholar] [CrossRef]
- Liu, Z.; Jian, Y.; Chen, Y.; Kistler, H.C.; He, P.; Ma, Z.; Yin, Y. A phosphorylated transcription factor regulates sterol biosynthesis in Fusarium graminearum. Nat. Commun. 2019, 10, 1228. [Google Scholar] [CrossRef]
- Maguire, S.L.; Wang, C.; Holland, L.M.; Brunel, F.; Neuvéglise, C.; Nicaud, J.M.; Zavrel, M.; White, T.C.; Wolfe, K.H.; Butler, G. Zinc finger transcription factors displaced SREBP proteins as the major sterol regulators during Saccharomycotina evolution. PLoS Genet. 2014, 10, e1004076. [Google Scholar] [CrossRef] [PubMed]
- Bien, C.M.; Espenshade, P.J. Sterol regulatory element binding proteins in fungi: Hypoxic transcription factors linked to pathogenesis. Eukaryot. Cell 2010, 9, 352–359. [Google Scholar] [CrossRef]
- Chauhan, N.; Inglis, D.; Roman, E.; Pla, J.; Li, D.; Calera, J.A.; Calderone, R. Candida albicans response regulator gene SSK1 regulates a subset of genes whose functions are associated with cell wall biosynthesis and adaptation to oxidative stress. Eukaryot. Cell 2003, 2, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.; Kruppa, M.; Calderone, R. The Ssk1p response regulator and Chk1p histidine kinase mutants of Candida albicans are hypersensitive to fluconazole and voriconazole. Antimicrob. Agents Chemother. 2007, 51, 3747–3751. [Google Scholar] [CrossRef]
- Day, A.M.; McNiff, M.M.; da Silva Dantas, A.; Gow, N.A.R.; Quinn, J. Hog1 regulates stress tolerance and virulence in the emerging fungal pathogen Candida auris. mSphere 2018, 3, e00506-18. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.J.; Yu, Y.M.; Kim, G.B.; Lee, G.W.; Maeng, P.J.; Kim, S.; Floyd, A.; Heitman, J.; Bahn, Y.S. Remodeling of global transcription patterns of Cryptococcus neoformans genes mediated by the stress-activated HOG signaling pathways. Eukaryot. Cell 2009, 8, 1197–1217. [Google Scholar] [CrossRef]
- Córdova-Alcántara, I.M.; Venegas-Cortés, D.L.; Martínez-Rivera, M.Á.; Pérez, N.O.; Rodriguez-Tovar, A.V. Biofilm characterization of Fusarium solani keratitis isolate: Increased resistance to antifungals and UV light. J. Microbiol. 2019, 57, 485–497. [Google Scholar] [CrossRef]
- Bispo, P.J.; Haas, W.; Gilmore, M.S. Biofilms in infections of the eye. Pathogens 2015, 4, 111–136. [Google Scholar] [CrossRef]
- Montgomery, M.L.; Fuller, K.K. Experimental models for fungal keratitis: An overview of principles and protocols. Cells 2020, 9, 1713. [Google Scholar] [CrossRef]
- Ponce-Angulo, D.G.; Bautista-Hernández, L.A.; Calvillo-Medina, R.P.; Castro-Tecorral, F.I.; Aparicio-Ozores, G.; López-Villegas, E.O.; Ribas-Aparicio, R.M.; Bautista-de Lucio, V.M. Microscopic characterization of biofilm in mixed keratitis in a novel murine model. Microb. Pathog. 2020, 140, 103953. [Google Scholar] [CrossRef]
- Wu, T.G.; Keasler, V.V.; Mitchell, B.M.; Wilhelmus, K.R. Immunosuppression affects the severity of experimental Fusarium solani keratitis. J. Infect. Dis. 2004, 190, 192–198. [Google Scholar] [CrossRef]
- Forster, R.K.; Rebell, G. Animal model of Fusarium solani keratitis. Am. J. Ophthalmol. 1975, 79, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Calvillo-Medina, R.P.; Reyes-Grajeda, J.P.; Barba-Escoto, L.; Bautista-Hernandez, L.A.; Campos-Guillén, J.; Jones, G.H.; Bautista-de Lucio, V.M. Proteome analysis of biofilm produced by a Fusarium falciforme keratitis infectious agent. Microb. Pathog. 2019, 130, 232–241. [Google Scholar] [CrossRef]
- Zhang, R.; Wiederhold, N.; Calderone, R.; Li, D. Biofilm formation in clinical isolates of Fusarium. J. Fungi 2024, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Chandra, J.; Kuhn, D.M.; Ghannoum, M.A. Mechanism of fluconazole resistance in Candida albicans biofilms: Phase-specific role of efflux pumps and membrane sterols. Infect. Immun. 2003, 71, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Ramage, G.; Rajendran, R.; Sherry, L.; Williams, C. Fungal biofilm resistance. Int. J. Microbiol. 2012, 2012, 528521. [Google Scholar] [CrossRef]
- Shay, R.; Wiegand, A.A.; Trail, F. Biofilm formation and structure in the filamentous fungus Fusarium graminearum, a plant pathogen. Microbiol. Spectr. 2022, 10, e0017122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Residues * of Missense Mutations in A. fumigatus CYP 51 Proteins | F. solani CYP51 | F. oxysporum CYP51 | F. fujikuroi CYP51 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | A | B | C | A | B | C | |
| A. fumigatus CYP51A | |||||||||
| G54I, Y121F | - | - | - | - | - | - | - | - | - |
| L98H | - | H | - | - | H | - | - | H | - |
| G138C | - | - | A | - | - | A | - | A | - |
| P216L, F219C | - | - | - | - | - | - | - | - | |
| M220I | L | I | - | L | V | - | L | - | V |
| N266I | - | T | H | - | T | H | - | H | T |
| T289A | - | A | A | - | A | A | - | A | A |
| Y431C, G432S | - | - | - | - | - | - | - | - | - |
| G448S | - | - | A | - | - | S | - | - | S |
| A. fumigatus CYP51B (Ref. [74]) | |||||||||
| G457S | - | - | - | - | - | - | - | - | - |
| A. flavus CYP51C (Ref. [75]) | |||||||||
| M54T | L | - | I | L | - | I | L | - | I |
| S240A ** | D | K | G | E | K | E | E | K | E |
| P419T | V | - | D | - | - | E | - | - | E |
| N423D | A | G | A | A | G | K | V | A | V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Amburgey-Crovetti, K.; Applebach, E.; Steen, T.Y.; Calderone, R. The Dual Pathogen Fusarium: Diseases, Incidence, Azole Resistance, and Biofilms. J. Fungi 2025, 11, 294. https://doi.org/10.3390/jof11040294
Li D, Amburgey-Crovetti K, Applebach E, Steen TY, Calderone R. The Dual Pathogen Fusarium: Diseases, Incidence, Azole Resistance, and Biofilms. Journal of Fungi. 2025; 11(4):294. https://doi.org/10.3390/jof11040294
Chicago/Turabian StyleLi, Dongmei, Kincer Amburgey-Crovetti, Emilie Applebach, Tomoko Y. Steen, and Richard Calderone. 2025. "The Dual Pathogen Fusarium: Diseases, Incidence, Azole Resistance, and Biofilms" Journal of Fungi 11, no. 4: 294. https://doi.org/10.3390/jof11040294
APA StyleLi, D., Amburgey-Crovetti, K., Applebach, E., Steen, T. Y., & Calderone, R. (2025). The Dual Pathogen Fusarium: Diseases, Incidence, Azole Resistance, and Biofilms. Journal of Fungi, 11(4), 294. https://doi.org/10.3390/jof11040294