
The Effects of Fungal Pathogen Infestation on Soil Microbial Communities for Morchella sextelata Cultivation on the Qinghai–Xizang Plateau

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sample Collection
2.2. Extraction, Sequencing, and Assembly of DNA from Soil Samples
2.3. Species Annotation
2.4. Functional Annotation
2.5. Statistical Analyses
2.6. Identification Methods for Pathogenic Fungi of Morchella
3. Results
3.1. Quality Control and Statistical Analysis of Metagenomic Sequencing Data
3.2. Metagenomic Sequencing Sequence Assembly and Preliminary Gene Prediction
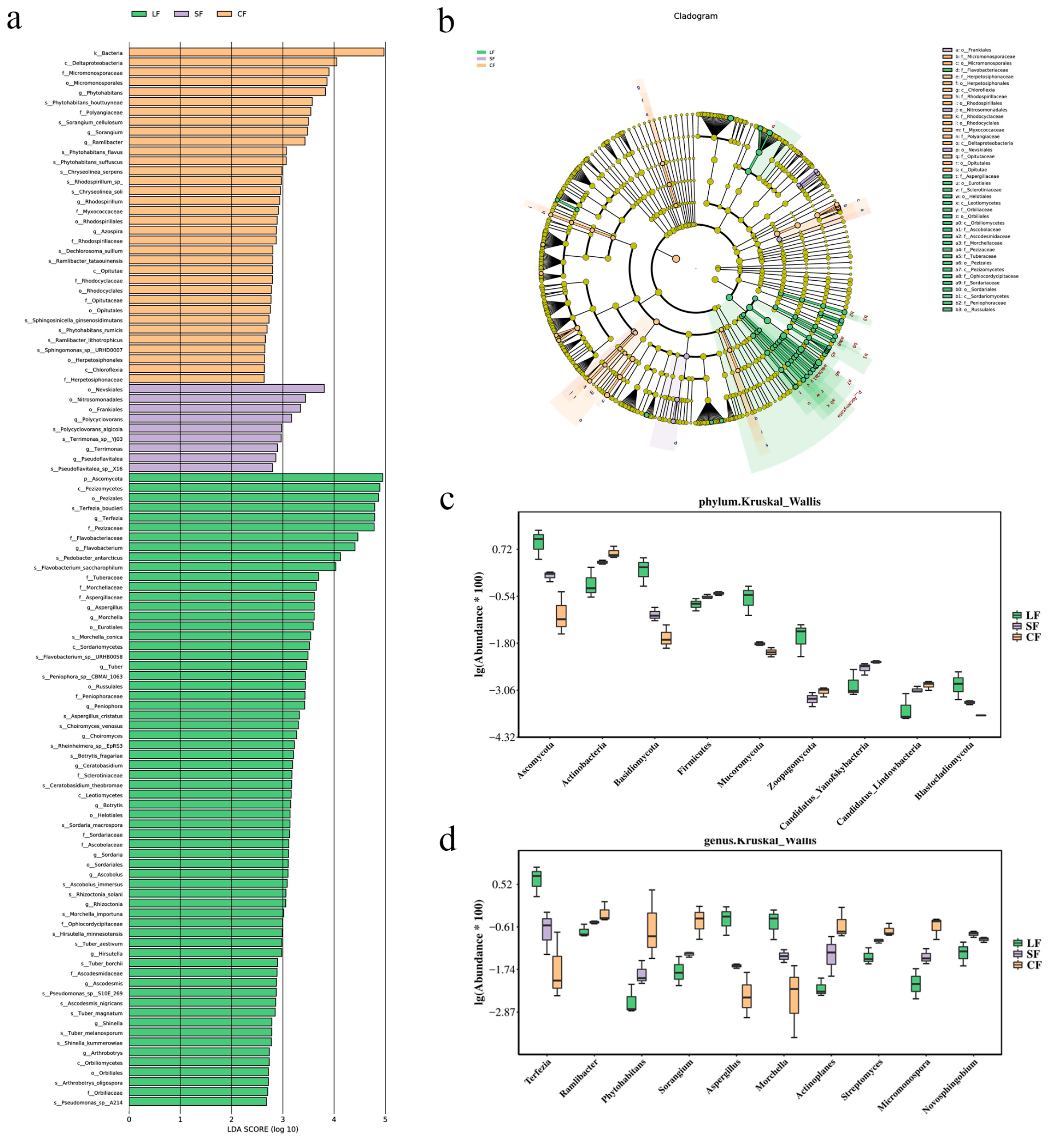
3.3. Species-Level Analysis of Each Sample
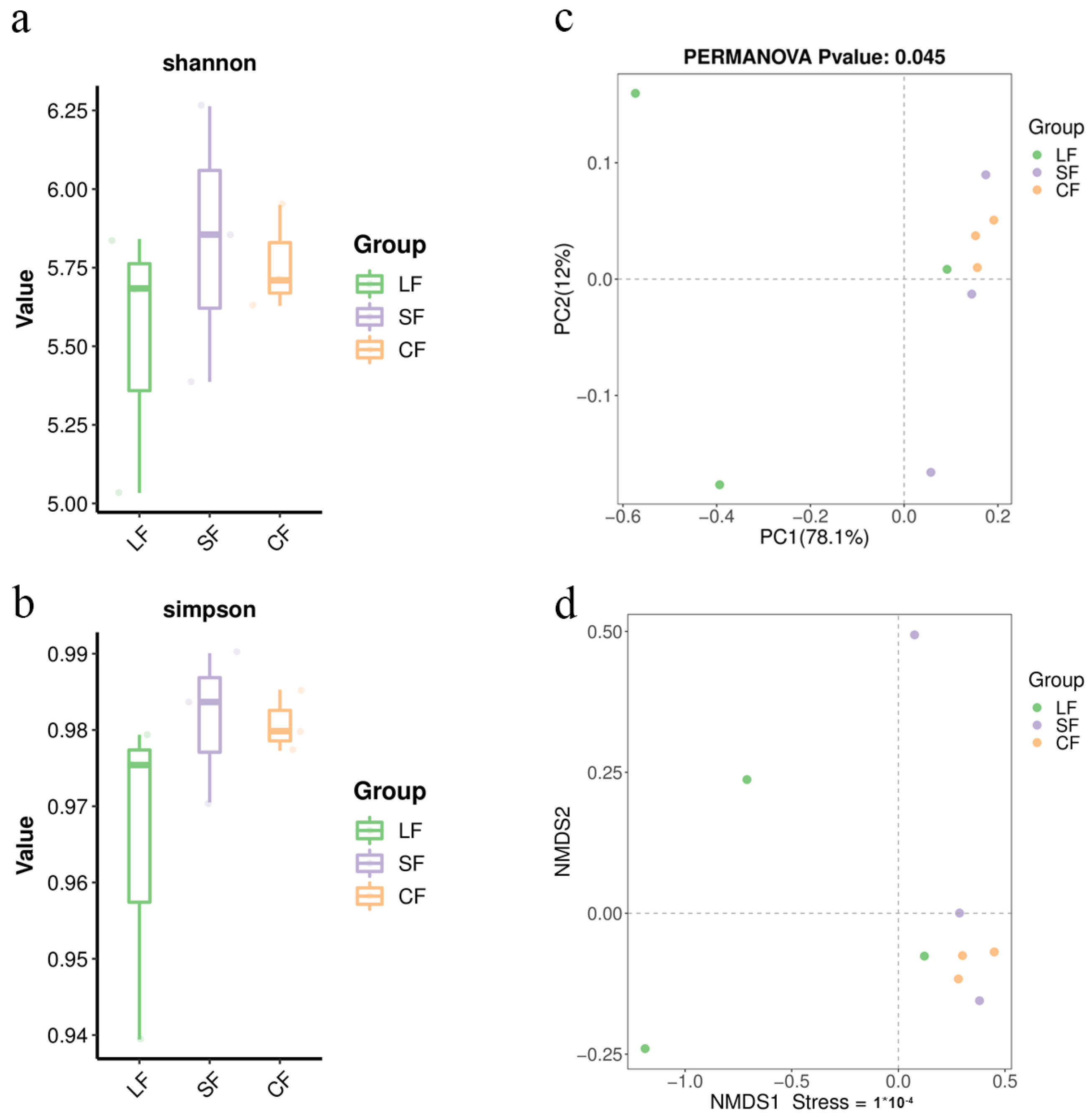
3.4. Analysis of Alpha Diversity Indices for Soil Samples
3.5. Analysis of Beta Diversity Indices for Soil Samples
3.5.1. PCoA Analysis of Soil Samples
3.5.2. NMDS Analysis of Soil Samples
3.6. Results of Non-Redundant Gene Functional Annotation
3.6.1. Statistics of Non-Redundant Gene Functional Annotation Results
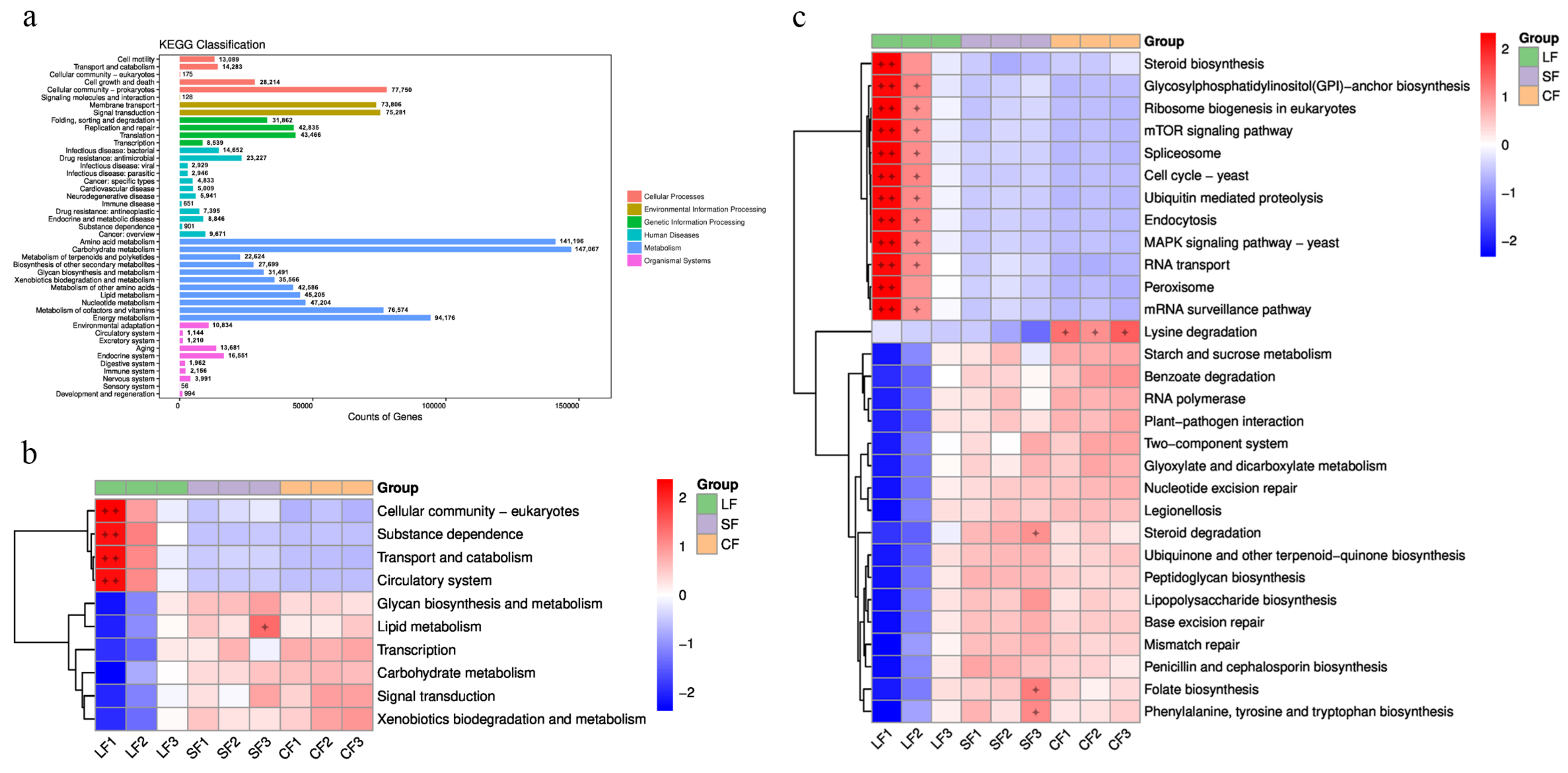
3.6.2. KEGG Analysis
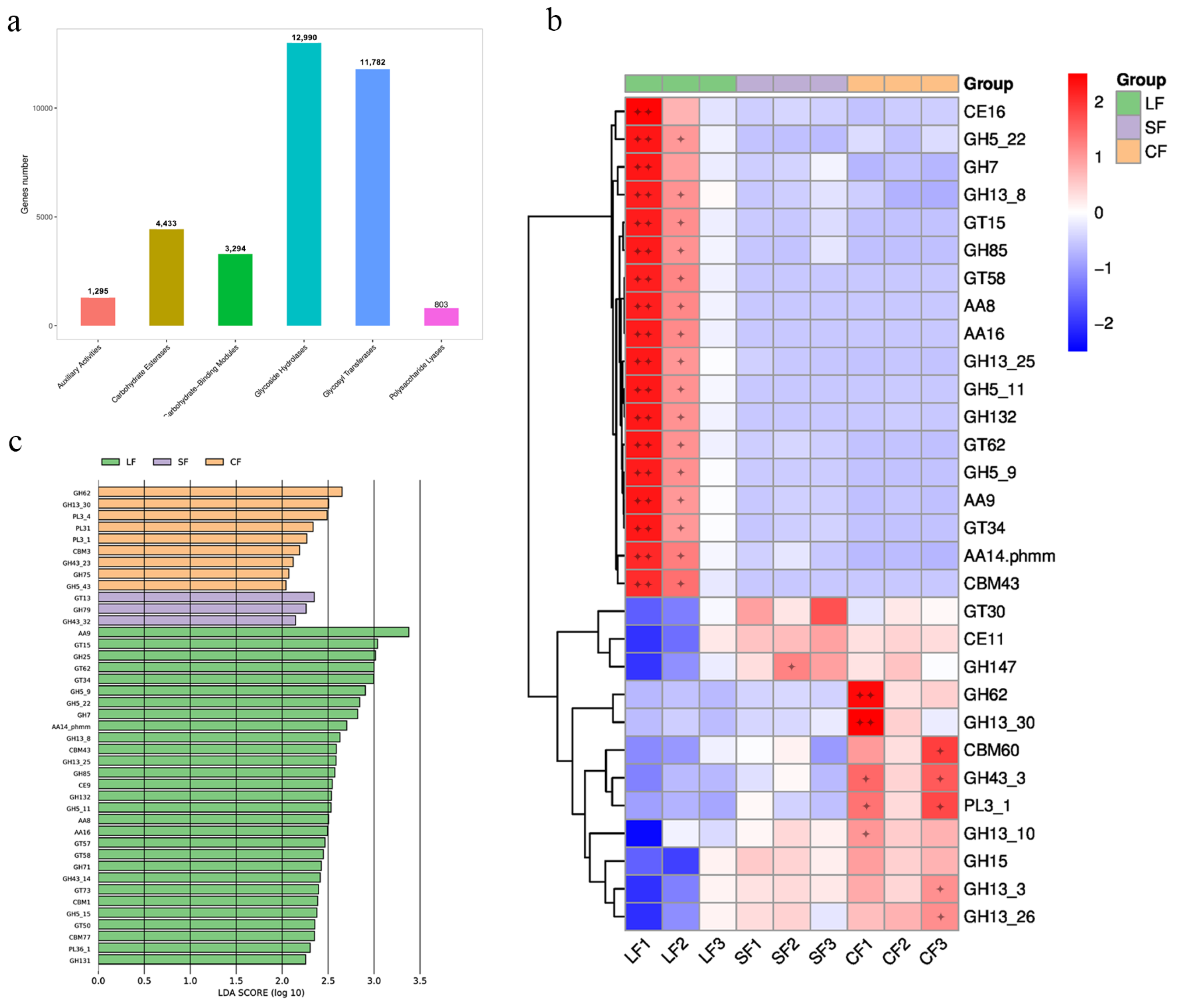
3.6.3. CAZY Analysis
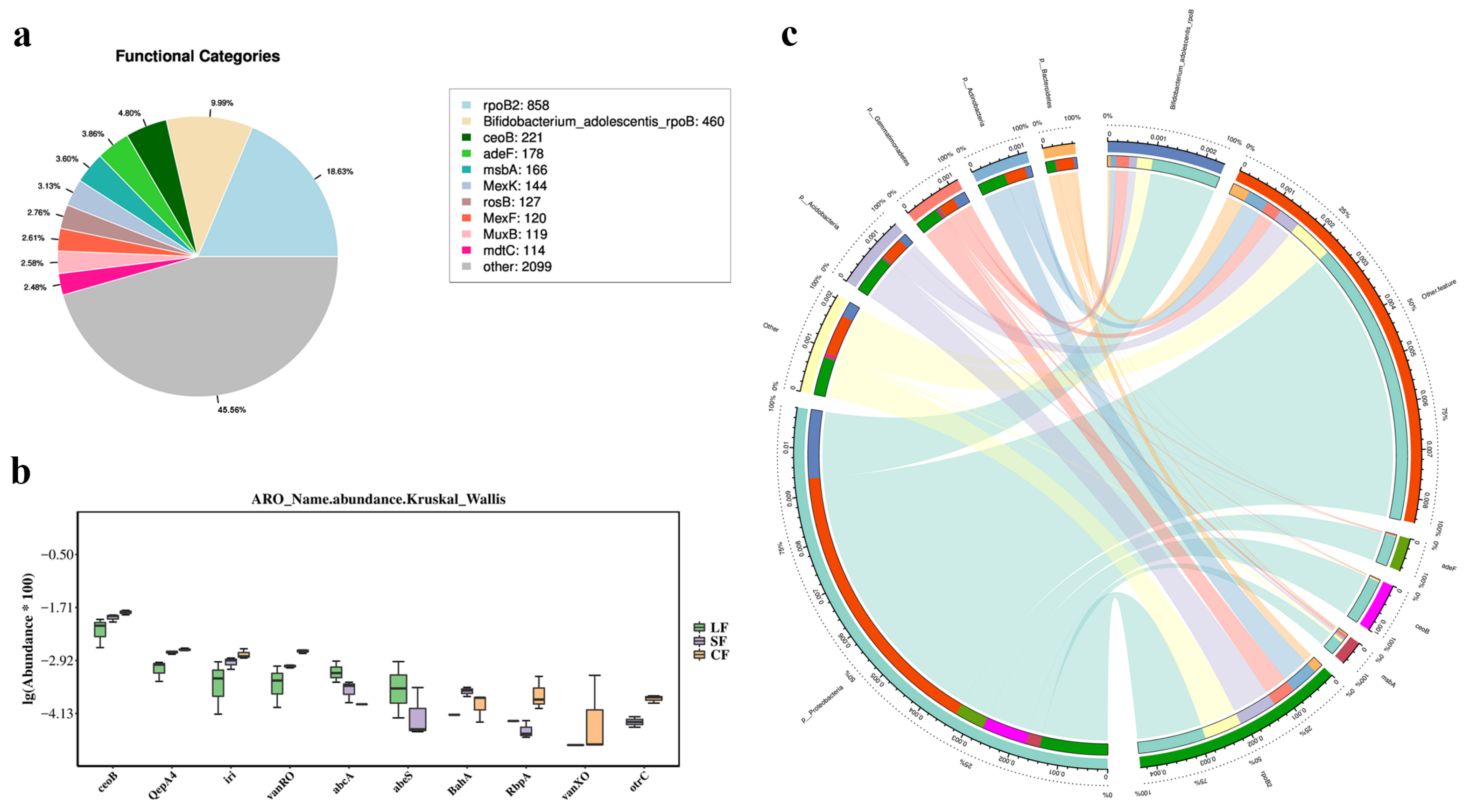
3.6.4. CARD Analysis
3.7. Molecular Biological Identification of Pathogenic Fungi
4. Discussion
4.1. Impact of Fungal Infection on the Soil Microbial Community of Morchella sextelata
4.2. Impact of Fungal Infection on Soil Functional Genes in Morchella sextelata
4.3. Impact of Fungal Infection on Soil Microbial Carbohydrate-Enzyme-Related Genes in Morchella sextelata
4.4. Effects of Fungal Infection on Resistance Gene Functions of Soil Microbiota in Morchella sextelata
4.5. Identification of Pathogenic Fungi in Morchella sextelata
5. Conclusions
- 1.
- Infection by P. lohjaoensis resulted in a lower microbial diversity in the Morchella soil community compared to the control group, while infection by T. gilva led to a higher microbial diversity compared to the control group.
- 2.
- Both the P. lohjaoensis and T. gilva infections altered the microbial community in the Morchella soil, with differences in the dominant phyla and genera observed in the different soil samples. After infection with P. lohjaoensis, the dominant phyla with relatively higher abundances included Proteobacteria (44%), Bacteroidetes (18%), and Ascomycota (15%). The dominant genera with relatively higher abundances were Pseudomonadaceae (8%), Terfezia (9%), and Pedobacter (5%). After infection with T. gilva, the dominant phyla with relatively higher abundances included Proteobacteria (46%), Acidobacteria (15%), and Bacteroidetes (12%). The dominant genera with relatively higher abundances were Hydrogenophaga (29%), Sphingomonas (3%), and Polaromonas (2%).
- 3.
- After infection by P. lohjaoensis, the microbial community structure in the soil where Morchella was growing underwent significant changes. However, after infection by T. gilva, the microbial community structure in the Morchella growth soil did not differ significantly from that of the healthy control group.
- 4.
- Fungal infection significantly altered microbial resistance functions in the soil where Morchella mushrooms were grown. Following infection by P. lohjaoensis, resistance functions were enriched in antibiotic permeability reduction. In contrast, infection by T. gilva led to an enrichment of resistance functions related to antibiotic target modification, target protection, and inactivation.
- 5.
- Using rDNA-ITS for strain identification, the pathogen in the LF group was identified as P. lohjaoensis, belonging to the order Pezizales, family Pezizaceae, and genus Peziza. The pathogen in the SF group was identified as T. gilva, belonging to the order Pezizales, family Pyronemataceae, and genus Tricharina.
- 6.
- Each sample in this study was replicated only three times, which may not be sufficiently representative of the overall sample. Therefore, the results may be limited by sample size and there is some statistical uncertainty. Future studies should consider increasing the sample size to improve the reliability and representativeness of the results.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tietel, Z.; Masaphy, S. True morels (Morchella)—Nutritional and phytochemical composition, health benefits and flavor: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhao, Q.; O’Donnell, K.; Rooney, A.P.; Yang, Z. Multigene molecular phylogenetics reveals true morels (Morchella) are especially species-rich in China. Fungal Genet. Biol. 2012, 49, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhao, Q.; Yang, Z. Diversity, evolutionary history and cultivation of morels: A review. Mycosystema 2014, 33, 183–197. [Google Scholar]
- Meng, F.; Liu, X.; Jia, L.; Song, Z.; Deng, P.; Fan, K. Optimization for the production of exopolysaccharides from Morchella esculenta SO-02 in submerged culture and its antioxidant activities in vitro. Carbohydr. Polym. 2010, 79, 700–704. [Google Scholar]
- Jacinto-Azevedo, B.; Valderrama, N.; Henríquez, K.; Aranda, M.; Aqueveque, P. Nutritional Value and Biological Properties of Chilean Wild and Commercial Edible Mushrooms. Food Chem. 2021, 356, 129651. [Google Scholar]
- Nitha, B.; Fijesh, P.V.; Janardhanan, K.K. Hepatoprotective activity of cultured mycelium of Morel mushroom, Morchella esculenta. Exp. Toxicol. Pathol. 2013, 65, 105–112. [Google Scholar]
- Hu, Q.; Liao, C.; Zou, Y. The current development status and prospect of rare edible fungi in China. Agric. Compr. Dev. China 2022, 4, 50–54. [Google Scholar]
- Liu, Q.; Ma, H.; Zhang, Y.; Dong, C. Artificial cultivation of true morels: Current state, issues and perspectives. Crit. Rev. Biotechnol. 2018, 38, 259–271. [Google Scholar]
- Masaphy, S. Biotechnology of morel mushrooms: Successful fruiting body formation and development in a soilless system. Biotechnol. Lett. 2010, 32, 1523–1527. [Google Scholar]
- Ower, R. Notes on the Development of the Morel Ascocarp: Morchella esculenta. Mycologia 1982, 74, 142–144. [Google Scholar]
- Ower, R.D.; Mills, G.L.; Malachowski, J.A. Cultivation of Morchella. U.S. Patent No. 4,594,809, 17 June 1986. [Google Scholar]
- He, P.; Liu, W.; Cai, Y.; He, X. Strain identification and phylogenetic analysis of cultivated and wild strains of Morchella belonging to Elata Clade in China. J. Zhengzhou Univ. Light Ind. Nat. Sci. 2015, 30, 26–29. [Google Scholar]
- Li, H.; Lin, J.; Fan, Y.; Mu, D. Technical regulations for green production of Morchella esculenta (morel mushroom) through facility cultivation in northwest region of China. Shaanxi J. Agric. Sci. 2024, 70, 64–66. [Google Scholar]
- Shu, L.; Qiu, Z.; Xu, H. Screening optimal light environment screening for facility cultivation of Morchella sextelata. J. Fungal Res. 2021, 19, 285–290. [Google Scholar]
- Zhou, S.; Sun, H.; Han, B.; Cen, X.; Jiang, J. Artificial cultivation technology of morels in small arched shed of forestland in north China. North. Hortic. 2021, 45, 165–167. [Google Scholar]
- Su, G.; Zou, X.; Xu, X.; Ye, W.; Su, H.; Ye, Q.; Yu, W. Production of Morchella sextelata strains and high–yield cultivation techniques in field facilities in western Zhejiang. Edible Med. Mushrooms 2024, 32, 72–76. [Google Scholar]
- He, X.; Wu, Y.; Wang, G.; Wang, J.; Wang, Y.; Li, X.; Zhao, M. The analysis and countermeasures for the large-scale failure of Morel mushroom production. Edible Fungi 2021, 43, 5–8. [Google Scholar]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, 245–249. [Google Scholar] [CrossRef]
- Wang, H.; Ju, F. Applications and perspectives of metagenomics in environmental microbiome research. Microbiol. China 2024, 51, 1814–1833. [Google Scholar]
- Zhang, C.; Jin, M.; Xu, Y.; Yang, C. Effects of maize continuous cropping on soil microbial community structure based on metagenomic analysis. Acta Prataculturae Sin. 2024, 33, 160–174. [Google Scholar]
- Pais, A.; Ristaino, J.; Whetten, R.; Xiang, Q. Metagenomic study reveals hidden relationships among fungal diversity, variation of plant disease, and genetic distance in Cornus florida (Cornaceae). Front. Plant Sci. 2024, 14, 1282188. [Google Scholar] [CrossRef]
- Xu, C.; Su, C.; Chen, Y.; Ma, J.; Wu, J.; Jiang, Y. Structure and functional heterogeneity of soil microbial communities in different farmland types on the Loess Plateau. J. Earth Environ. 2024, 15, 653–664. [Google Scholar]
- Wang, P.; Yang, Y.; Liu, D.; Cui, X.; Liu, Y. Application of Metagenomics in Plant Diseases Research. Biotechnol. Bull. 2020, 36, 146–154. [Google Scholar]
- Zhao, Y.; Cai, H.; Chen, W. Rational understanding of the development of Morchella industry. Edible Med. Mushrooms 2018, 26, 121–127. [Google Scholar]
- Guo, M.; Chen, K.; Wang, G.; Bian, Y. First report of stipe rot disease on Morchella importuna caused by Fusarium incarnatum—F. equiseti species complex in China. Plant Dis. 2016, 100, 2530. [Google Scholar]
- Lv, B.; Sun, Y.; Chen, Y.; Yu, H.; Mo, Q. First report of Lecanicillium aphanocladii causing rot of Morchella sextelata in China. Plant Dis. 2021, 106, 3202. [Google Scholar]
- Yu, M.; Yin, Q.; He, P. Isolation and identification of pathogen of morel white rot. North. Hortic. 2020, 7, 142–145. [Google Scholar]
- Zhang, Y.; Lu, Q.; Wang, Y.; Tu, C.; Liu, L.; Xie, Y.; Liu, J. Isolation and identification of Morchella pathogenic bacteria and determination of antibacterial activity of 22 essential oils. IOP Conf. Ser. Earth Environ. Sci. 2020, 615, 012098–012103. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, J.; Wang, D.; He, X.; Tang, J.; Chie, Y.; Wang, J.; Peng, W. A new stipe rot disease of the cultivated Morchella sextelata. Mycosystema 2021, 40, 2229–2243. [Google Scholar]
- Gao, Z.; Hua, R.; Sun, D.; Liu, S.; Yue, W.; Zhang, X.; Yu, S.; Li, J. Isolation and identification of two disease pathogens from Morchella spp. Edible Fungi China 2023, 42, 109–113. [Google Scholar]
- Zhang, X.; Yang, X.; Jiao, Z. Functional diversity analysis of rhizosphere soil microorganism of wide Morchella in Yily. J. Microbiol. 2019, 39, 83–89. [Google Scholar]
- Chen, C.; Li, Q.; Wang, J.; Fu, R.; Jin, X.; Xiong, C.; Lu, D. Effects of morel, Morchella sp. stipe rot disease occurrence on soil fungal community structure. J. Microbiol. 2018, 38, 39–45. [Google Scholar]
- Tan, Y.; Su, W.; Ren, L.; Fan, J.; Zhou, Z.; Yang, H. Effect of Paecilomyces penicillatus occurrence on fungal community Structure of Morchella rhizosphere soil. Acta Agric. Boreali-Occident. Sin. 2021, 30, 1227–1232. [Google Scholar]
- Xiong, C.; Li, X.; Li, Q.; Zheng, L. Bacteria community structure and diversity in Morchella colonies. J. Hunan Agric. Univ. 2015, 41, 428–434. [Google Scholar]
- Wang, S.; Liu, G.; Zhao, G. Investigation on resources and ecological environment Morchella species in the Yellow River flood plain of Heze city. Edible Fungi China 2008, 27, 12–14. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [PubMed]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar]
- Karlsson, F.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar]
- Xia, F.; Chen, X.; Guo, M.Y.; Bai, X.H.; Liu, Y.; Shen, G.R.; Li, Y.L.; Lin, J.; Zhou, X.W. High-throughput sequencing-based analysis of endogenetic fungal communities inhabiting the Chinese Cordyceps reveals unexpectedly high fungal diversity. Sci. Rep. 2016, 6, e3343. [Google Scholar]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar]
- Zhao, Q.; Xu, Z.; Cheng, Y.; Qi, S.; Hou, Z. Bionic cultivation of Morchella conica. Southwest China J. Agric. Sci. 2009, 22, 1690–1693. [Google Scholar]
- Zhu, D. Research progress in artificial cultivation of Morchella. Edible Fungi China 2008, 27, 3–5. [Google Scholar]
- Zhang, Z. Analysis of the rhizosphere and non-rhizosphere microbial communities of morel mushrooms. Edible Fungi China 1999, 18, 26–27. [Google Scholar]
- Kang, C.; Zeng, W.; Zheng, X.; Wang, W.; Yang, L.; Liu, Z.; Wang, J. Impacts of Morchella on physicochemical properties, microbial diversity and community structure of overburden soil layer. Jiangsu Agric. Sci. 2023, 51, 221–228. [Google Scholar]
- Zhang, T.; Huang, X.; Xu, Y.; Li, J. Study on microbial community structure and diversity of Morchella rhizosphere soil. J. Hebei Agric. Univ. 2018, 41, 38–43. [Google Scholar]
- Orlofsky, E.; Zabari, L.; Bonito, G.; Masaphy, S. Changes in soil bacteria functional ecology associated with Morchella rufobrunnea fruiting in a natural habitat. Environ. Microbiol. 2021, 23, 6651–6662. [Google Scholar]
- Tan, H.; Yu, Y.; Tang, J.; Liu, T.-T.; Miao, R.-Y.; Huang, Z.-Q.; Martin, F.M.; Peng, W.-H. Build your own mushroom soil: Microbiota succession and nutritional accumulation in semi-synthetic substratum drive the fructification of a soil-saprotrophic morel. Front. Microbiol. 2021, 12, 656656. [Google Scholar]
- Zhang, C.; Shi, X.; Zhang, J.; Zhang, Y.; Wang, W. Dynamics of soil microbiome throughout the cultivation life cycle of morel (Morchella sextelata). Front. Microbiol. 2023, 14, 979835. [Google Scholar]
- Benucci, G.M.N.; Longley, R.; Zhang, P.; Zhao, Q.; Bonito, G.; Yu, F.-Q. Microbial communities associated with the black morel Morchella sextelata cultivated in greenhouses. PeerJ 2019, 2019, e7744. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y. Research progresson infectious and competitive mycemial diseases of edible mushrooms. Acta Edulis Fungi 2013, 20, 1–7. [Google Scholar]
- Liu, Z.; Li, D.; Leo, S.F.; Shen, H.; Azu, O.B.; Fu, Y. Major pathogenic fungi and bacteria in edible fungi. J. Fungal Res. 2018, 16, 158–163. [Google Scholar]
- Liu, W.; Cai, Y.; He, P. Occurrence and Control of Pests and Diseases in Field Cultivation of Morchella Mushrooms. Acta Edulis Fungi 2019, 26, 128–134. [Google Scholar]
- He, P.; Li, C.; Cai, Y.; Zhang, Y.; Bian, Y.; Liu, W. First report of pileus rot disease on cultivated Morchella importuna caused by Diploöspora longispora in China. J. Gen. Plant Pathol. 2018, 84, 65–69. [Google Scholar] [CrossRef]
- Tong, L.; Xu, A.; Bao, D.; Wang, W.; Li, F.; Tang, L. Transcriptome mining of genes involved in drought stress response in Morchella sextelata. Acta Edulis Fungi 2024, 31, 26–33. [Google Scholar]
- Li, Y.; Li, Y.; Xu, L. Research on the effects of environmental factors and nutritional factors on growth and development of edible fungi. Anhui Agric. Sci. Bull. 2022, 28, 39–40. [Google Scholar]
- Wang, Y.; Li, Y.; Xu, H.; Cao, X. Effects of different carbon and nitrogen sources on mycelial growth and sclerotia formation of Morchella spp. North. Hortic. 2021, 45, 125–131. [Google Scholar]
- Wei, S.; Xu, M.; Yang, Q.; Zhang, J. Changes of metabolites in exogenous nutrient bags of Morchella esculenta cultivation. Acta Edulis Fungi 2023, 30, 18–30. [Google Scholar]
- He, S.; Zhao, K.; Ma, L.; Yang, J.; Chang, Y. Effects of different cultivation material formulas on the growth and quality of Morchella spp. Saudi J. Biol. Sci. 2018, 25, 719–723. [Google Scholar] [CrossRef]
- Vajna, B.; Szili, D.; Nagy, A.; Márialigeti, K. An improved sequence-aided T-RFLP analysis of bacterial succession during oyster mushroom substrate preparation. Microb. Ecol. 2012, 64, 702–713. [Google Scholar] [PubMed]
- Zhang, X.; Zhong, Y.; Yang, S.; Zhang, W.; Xu, M.; Ma, A.; Zhuang, G.; Chen, G.; Liu, W. Diversity and dynamics of the microbial community on decomposing wheat straw during mushroom compost production. Bioresour. Technol. 2014, 170, 183–195. [Google Scholar] [PubMed]
- Gohar, D.; Põldmaa, K.; Tedersoo, L.; Aslani, F.; Furneaux, B.; Henkel, T.W.; Saar, I.; Smith, M.E.; Bahram, M. Global diversity and distribution of mushroom-inhabiting bacteria. Environ. Microbiol. Rep. 2022, 14, 254–264. [Google Scholar]
- Huang, P.; Ding, M.; Zhang, H.; Xu, H.; Zou, T.; Zhang, Y.; Zeng, H. Responses of soil microbial structure and carbon cycling functions to degradation and artificial restoration of alpine meadows. Chin. J. Ecol. 2024, 43, 1691–1702. [Google Scholar]
- Ma, Y.; Sun, D.; Yue, H.; Xin, J.; Liu, N.; Cao, Z. Identification, Heterologous Expression and Functional Analysis of a GH61 Family Glycoside Hydrolase from Setosphaeria turcica with the Assisting Function in Degrading Cellulose. Biotechnol. Bull. 2023, 39, 124–135. [Google Scholar]
- Li, J.; Liu, C.; Shang, L.; Yang, Z.; Luo, X.; Liu, S. Tentative exploration on associated microorganism of Morchella sextelata and Peziza spp. Edible Fungi China 2020, 39, 17–19. [Google Scholar]
- Forsberg, K.J.; Reyes, A.; Wang, B.; Selleck, E.M.; Sommer, M.O.A.; Dantas, G. The shared antibiotic resistome of soil bacteria and human pathogens. Science 2012, 337, 1107–1111. [Google Scholar]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar]
- Nesme, J.; Cécillon, S.; Delmont, T.O.; Monier, J.M.; Vogel, T.M.; Simonet, P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr. Biol. 2014, 24, 1096–1100. [Google Scholar]
- D’costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R. Antibiotic resistance is ancient. Nature 2011, 477, 457. [Google Scholar] [CrossRef]
- Zhuang, W. Some new species and new records of discomycetes in China. Mycotaxon 2002, 21, 475–479. [Google Scholar]
- Bai, X.; Zhang, E.; Wu, J.; Ma, D.; Zhang, B.; Zhang, C.; Tian, F.; Zhao, H.; Wang, B. Effects of different modified materials on soil fungal community structure in saline-alkali soil. Environ. Sci. 2024, 45, 3562–3570. [Google Scholar]
- Tian, H.; Wang, X.; Li, X. Isolation, identification of a dark septate endophytes from Ulmus pumila L. and function examination of antagonism to plant pathogenic fungi. J. Hunan Agric. Univ. (Nat. Sci.) 2022, 48, 416–422. [Google Scholar]
- Yang, Z.; Fan, B.; Zhang, Q.; Liu, Y.; Pan, J.; Feng, H. Isolation and identification of foliar endophytic fungi of Picea crassifolia in the Qilian Mountains. Biotic Res. 2024, 46, 39–50. [Google Scholar]
- Mbilu, M.; Wanyoike, W.; Kangogo, M.; Bii, C.; Agnes, M.; Kihia, C. Isolation and characterization of endophytic fungi from medicinalplant Warburgia ugandensis. J. Biol. Agric. Healthc. 2018, 8, 57–66. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.-C.; Wu, B.-C.; Luo, C.-Y.; Sa, W.; Wang, L.; Li, Z.-H.; Shang, Q.-H. The Effects of Fungal Pathogen Infestation on Soil Microbial Communities for Morchella sextelata Cultivation on the Qinghai–Xizang Plateau. J. Fungi 2025, 11, 264. https://doi.org/10.3390/jof11040264
Guo M-C, Wu B-C, Luo C-Y, Sa W, Wang L, Li Z-H, Shang Q-H. The Effects of Fungal Pathogen Infestation on Soil Microbial Communities for Morchella sextelata Cultivation on the Qinghai–Xizang Plateau. Journal of Fungi. 2025; 11(4):264. https://doi.org/10.3390/jof11040264
Chicago/Turabian StyleGuo, Ming-Chen, Bo-Chun Wu, Cai-Yun Luo, Wei Sa, Le Wang, Zhong-Hu Li, and Qian-Han Shang. 2025. "The Effects of Fungal Pathogen Infestation on Soil Microbial Communities for Morchella sextelata Cultivation on the Qinghai–Xizang Plateau" Journal of Fungi 11, no. 4: 264. https://doi.org/10.3390/jof11040264
APA StyleGuo, M.-C., Wu, B.-C., Luo, C.-Y., Sa, W., Wang, L., Li, Z.-H., & Shang, Q.-H. (2025). The Effects of Fungal Pathogen Infestation on Soil Microbial Communities for Morchella sextelata Cultivation on the Qinghai–Xizang Plateau. Journal of Fungi, 11(4), 264. https://doi.org/10.3390/jof11040264