


The Three-Dimensional Structure of the Genome of the Dark Septate Endophyte Exophiala tremulae and Its Symbiosis Effect on Alpine Meadow Plant Growth


Abstract
1. Introduction
2. Experimental Materials and Methods
2.1. Culture and Morphological Observation of E. tremulae
2.2. Symbiosis Culture of E. tremulae and M. sativa Seedlings and Growth Investigation
2.3. Extraction of DNA and RNA from the Hyphae of E. tremulae
2.4. Construction of PacBio HiFi Library
2.5. Construction of Hi-C Libraries
2.6. Genome Assembly with HiFi Reads
2.7. Chromosome Assignment Using Hi-C Technology
2.8. Assessment of Assembly Quality
2.9. Genome Annotation
2.9.1. Repeat Sequence Annotation
2.9.2. Gene Annotation
2.9.3. Functional Annotations
2.9.4. Annotation of Non-Coding RNA Genes
2.10. Comparative Genome Analysis
2.10.1. Gene Family Identification
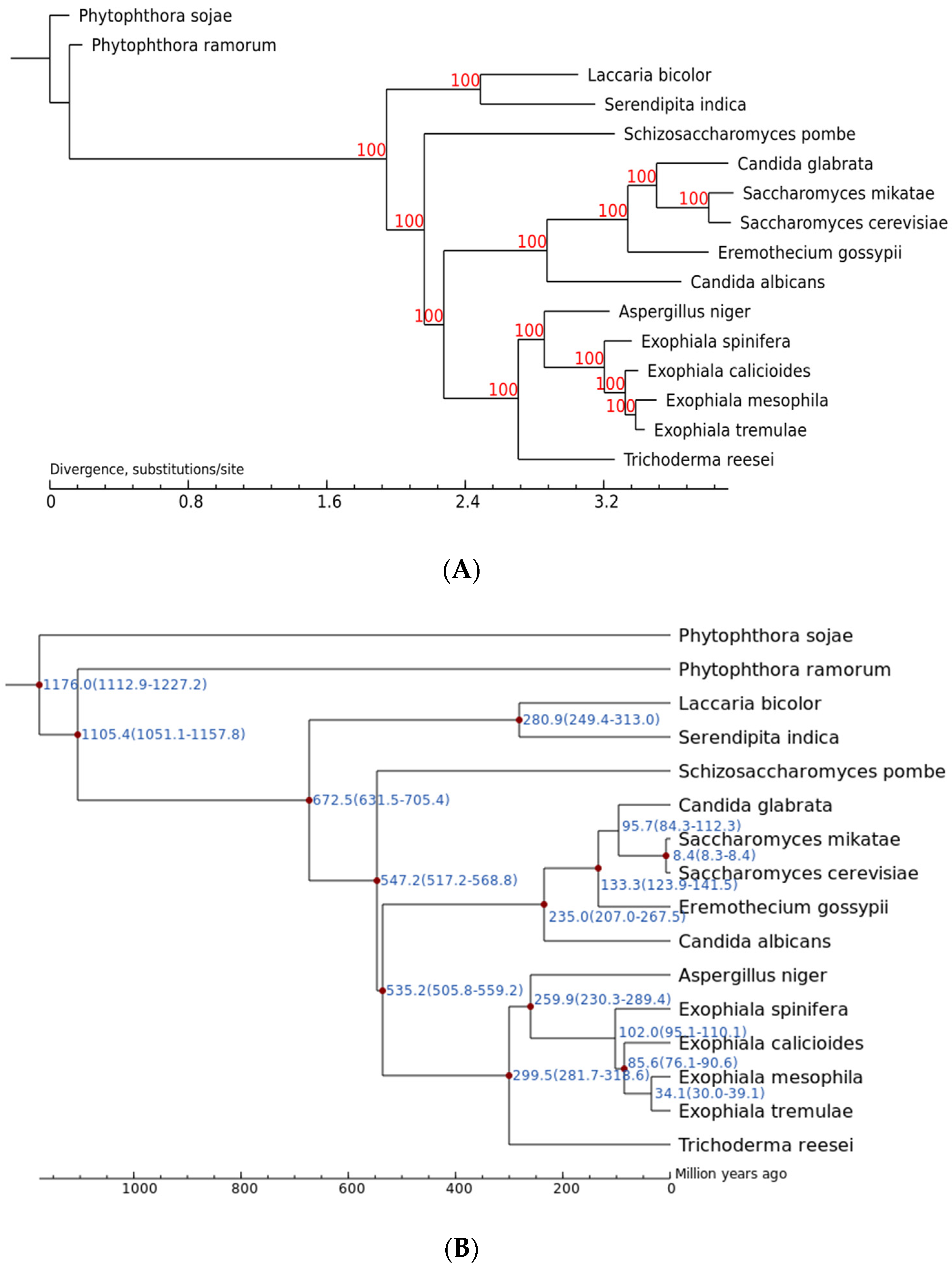
2.10.2. Phylogenetic Analysis
2.10.3. Gene Family Expansion and Contraction Analysis
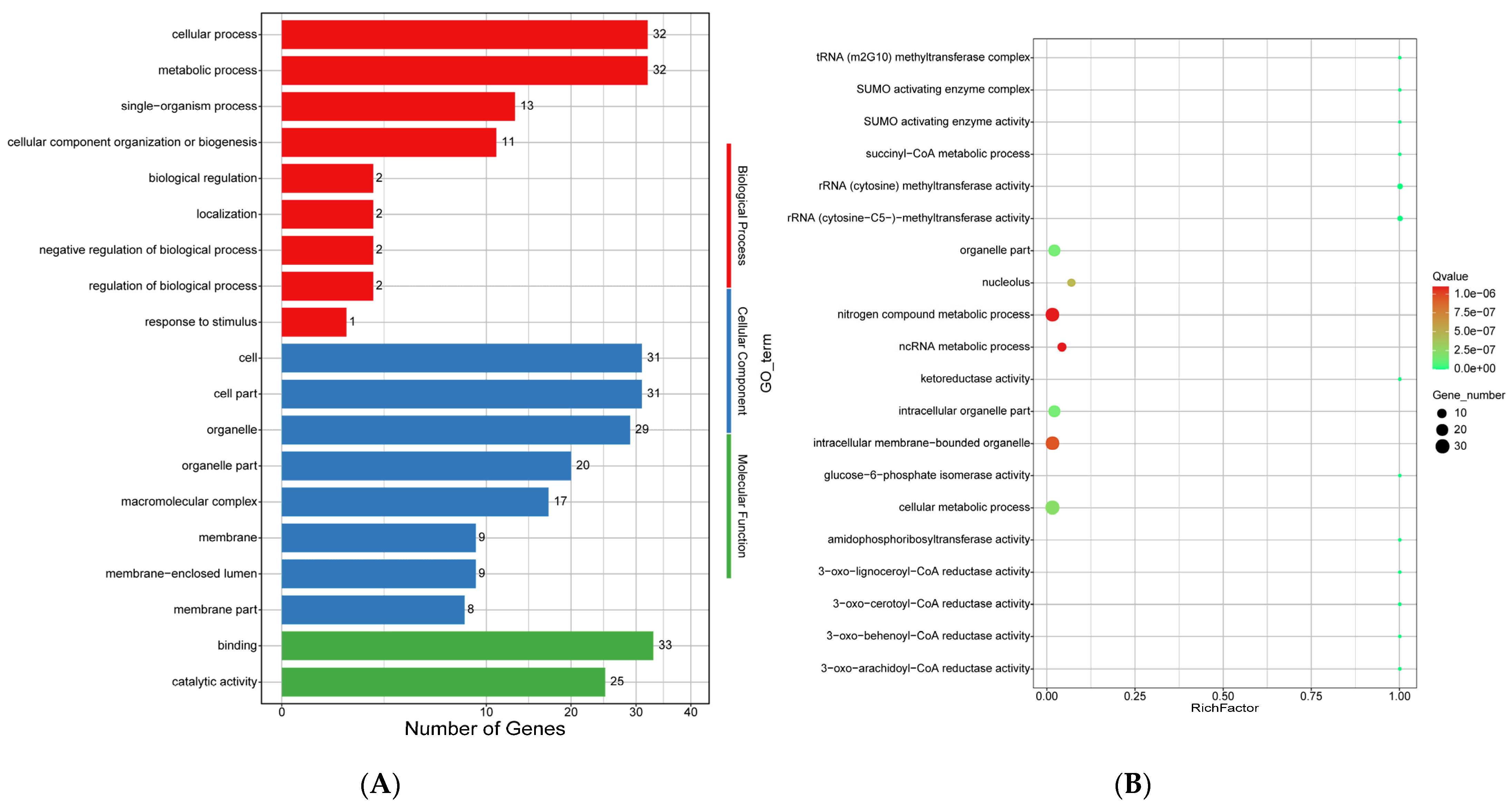
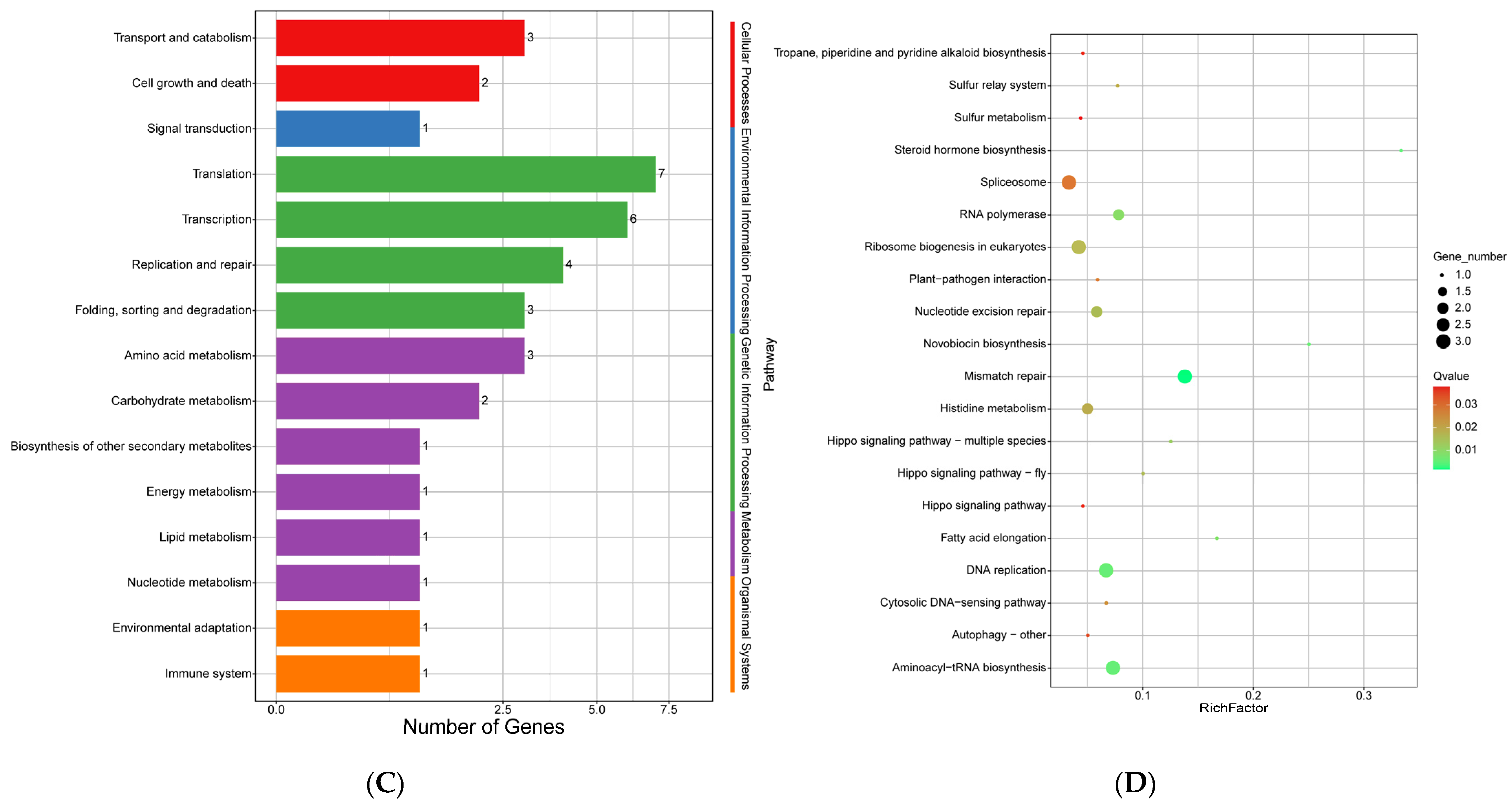
2.10.4. Analysis of Positively Selected Genes
2.11. Statistical Analysis
3. Results
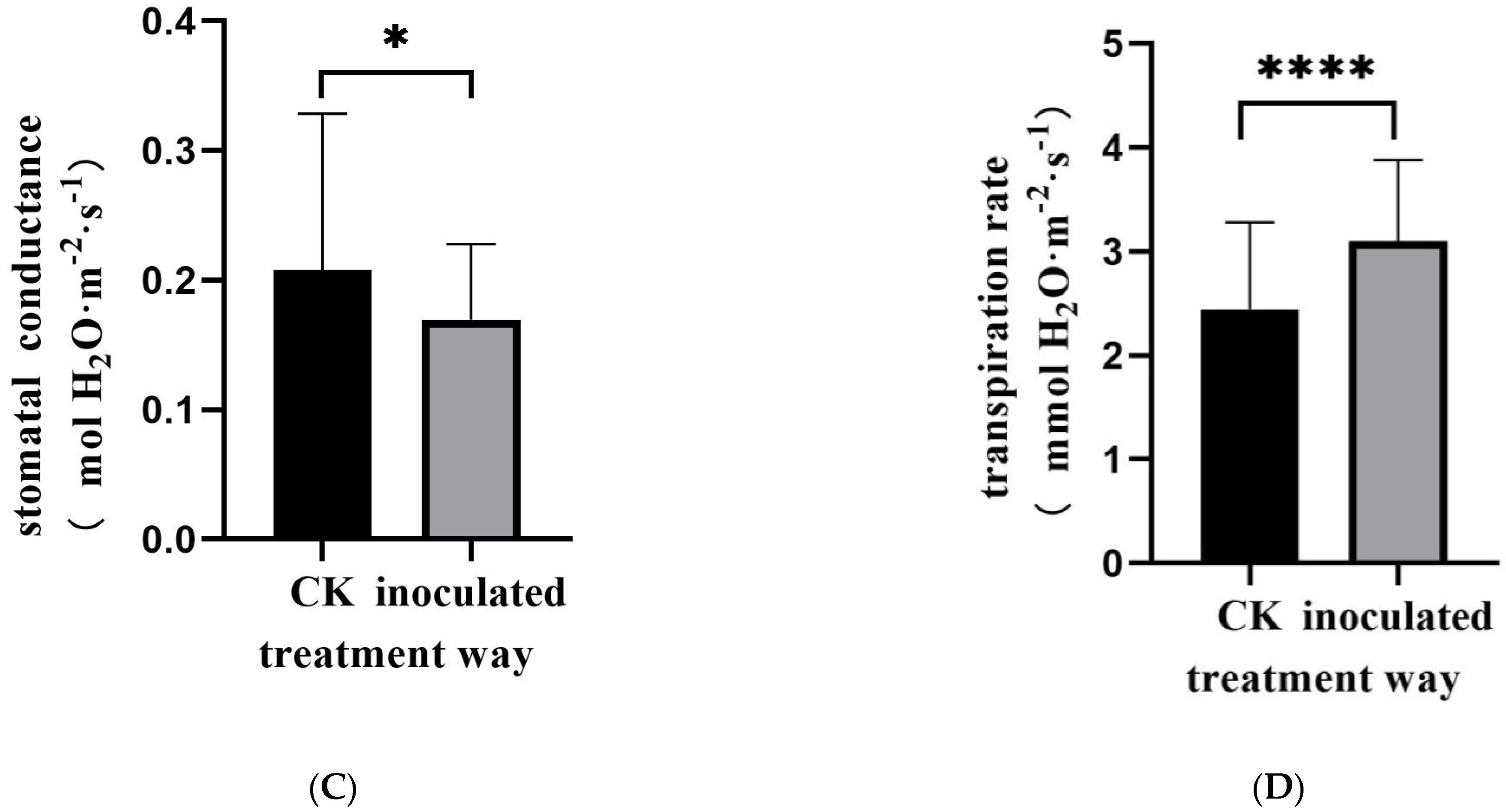
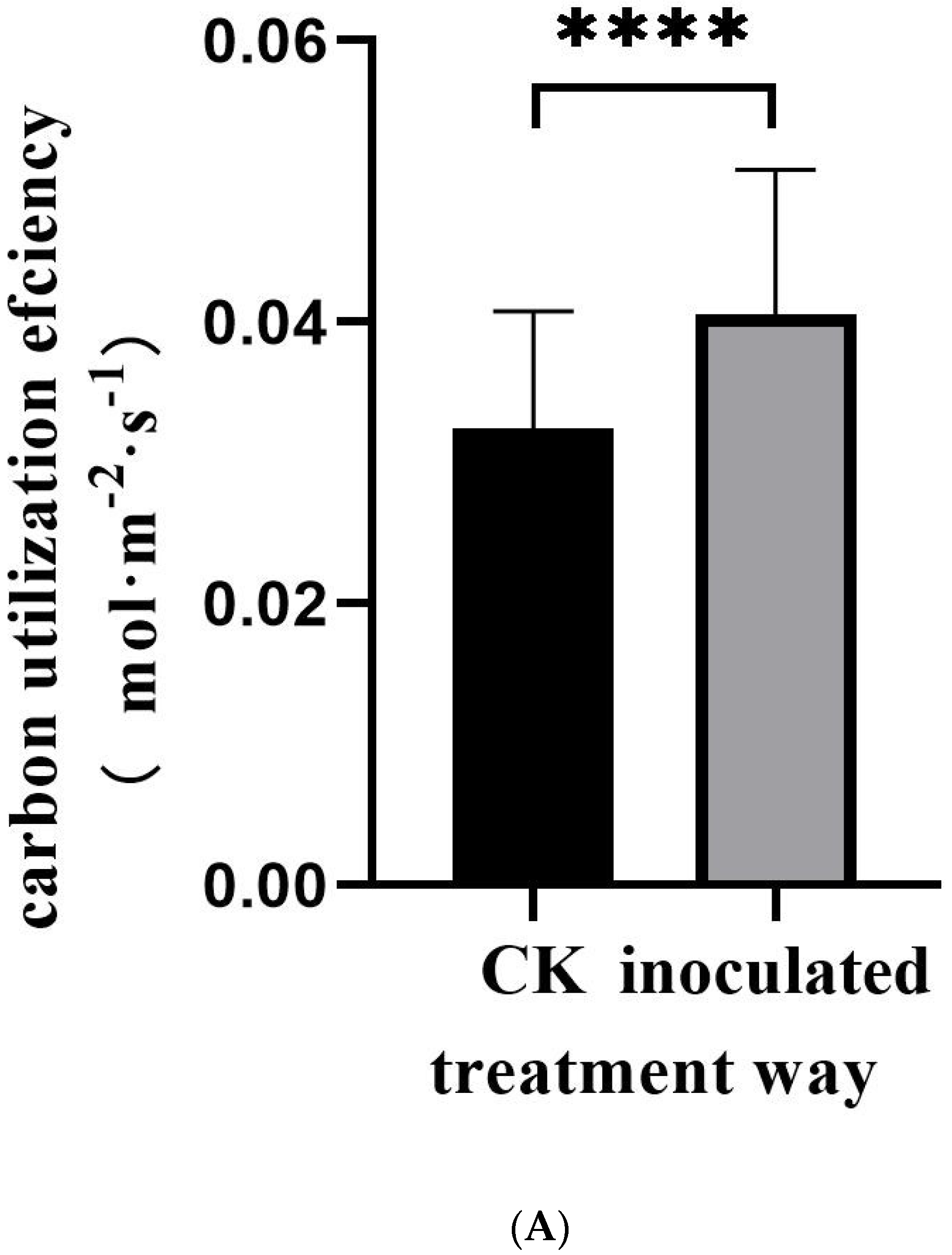
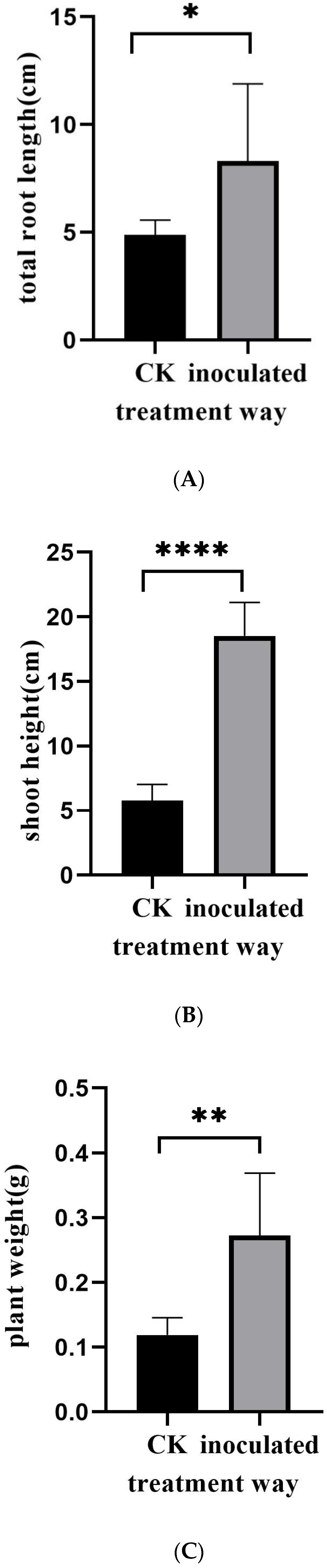
3.1. Morphological Observation of E. tremulae and Its Symbiosis Effect on Growth of M. sativa Seedlings
3.2. Chromosome-Level Assembly of the Genome of E. tremulae
3.3. Genome Annotation
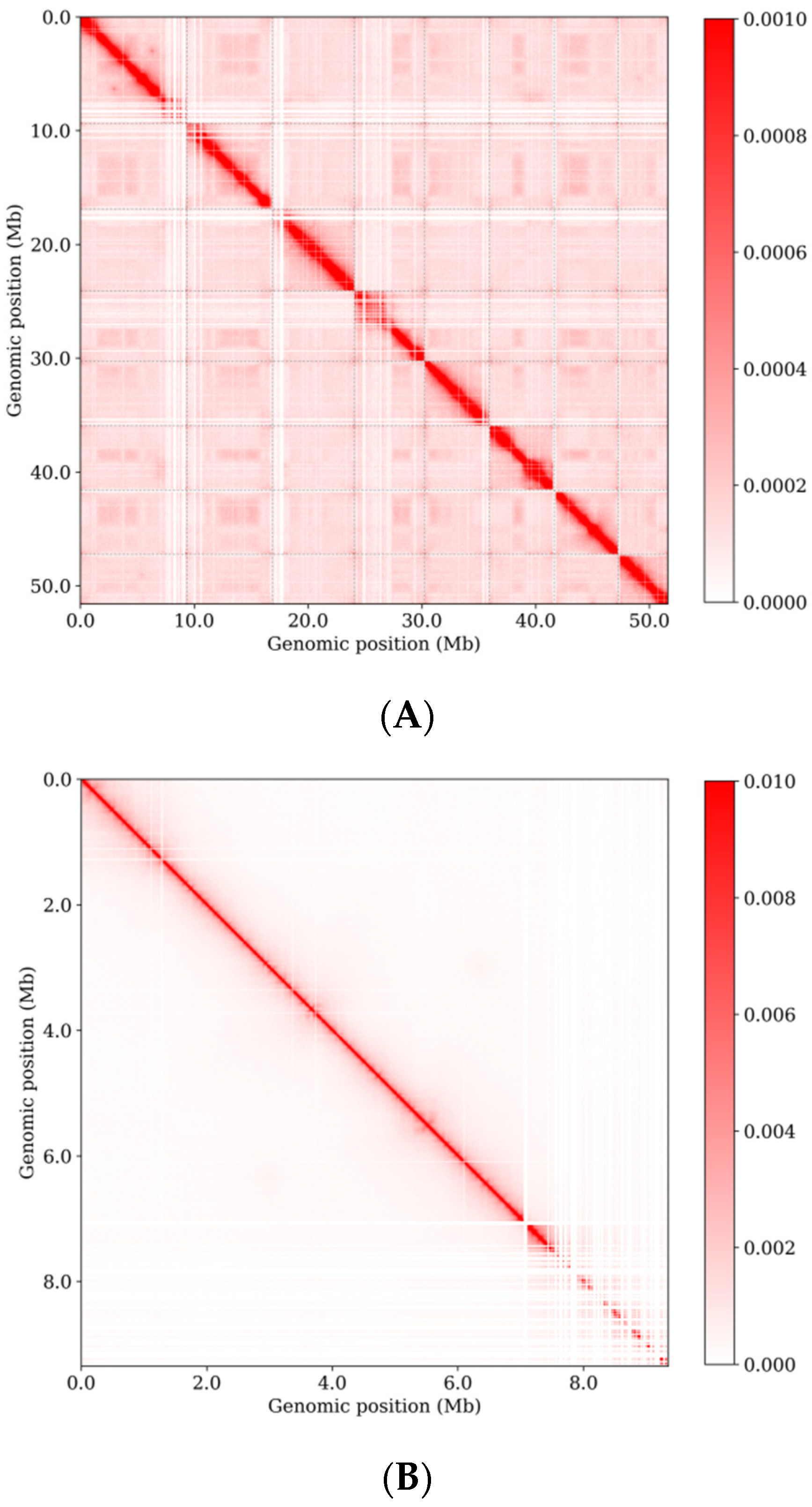
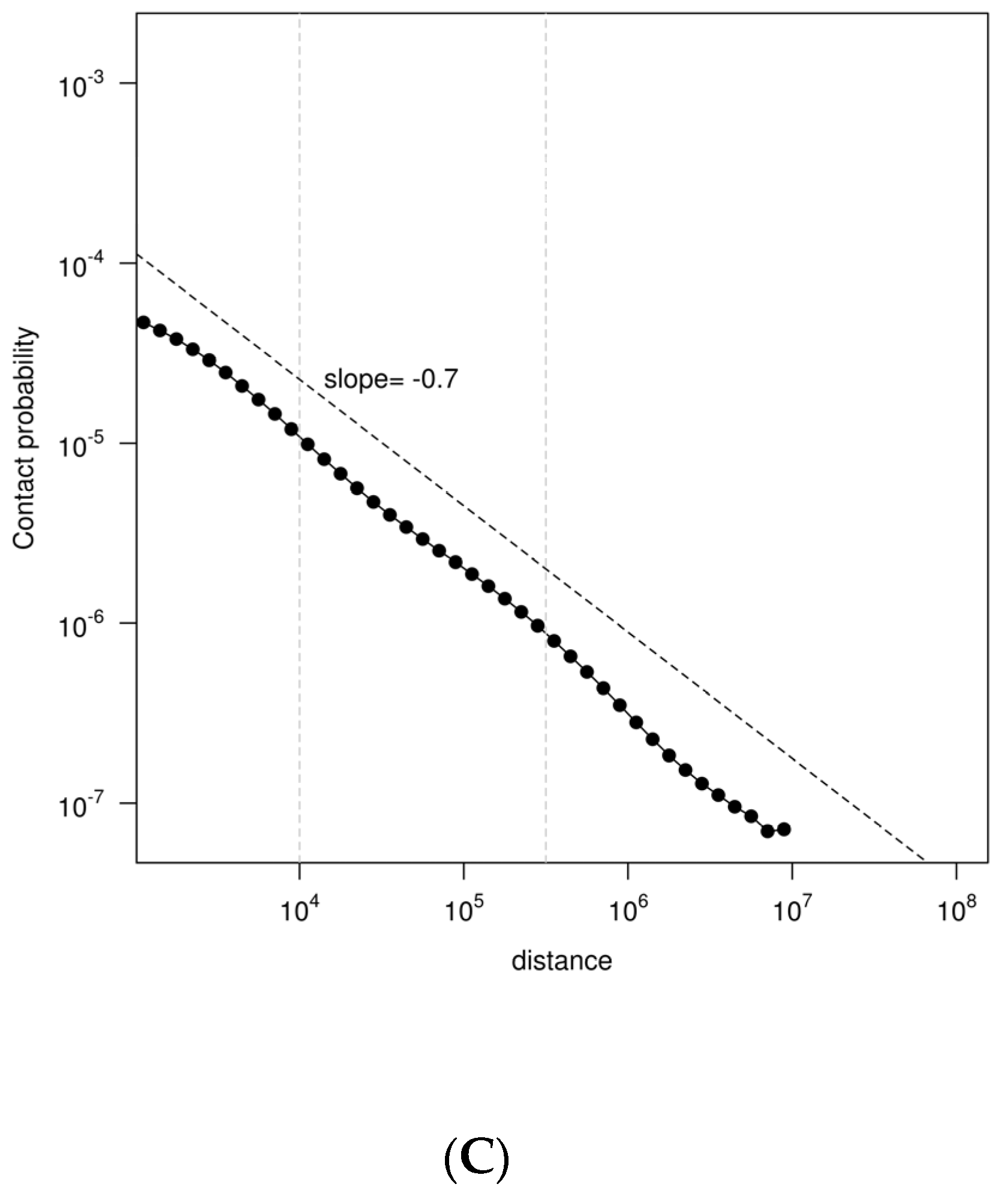
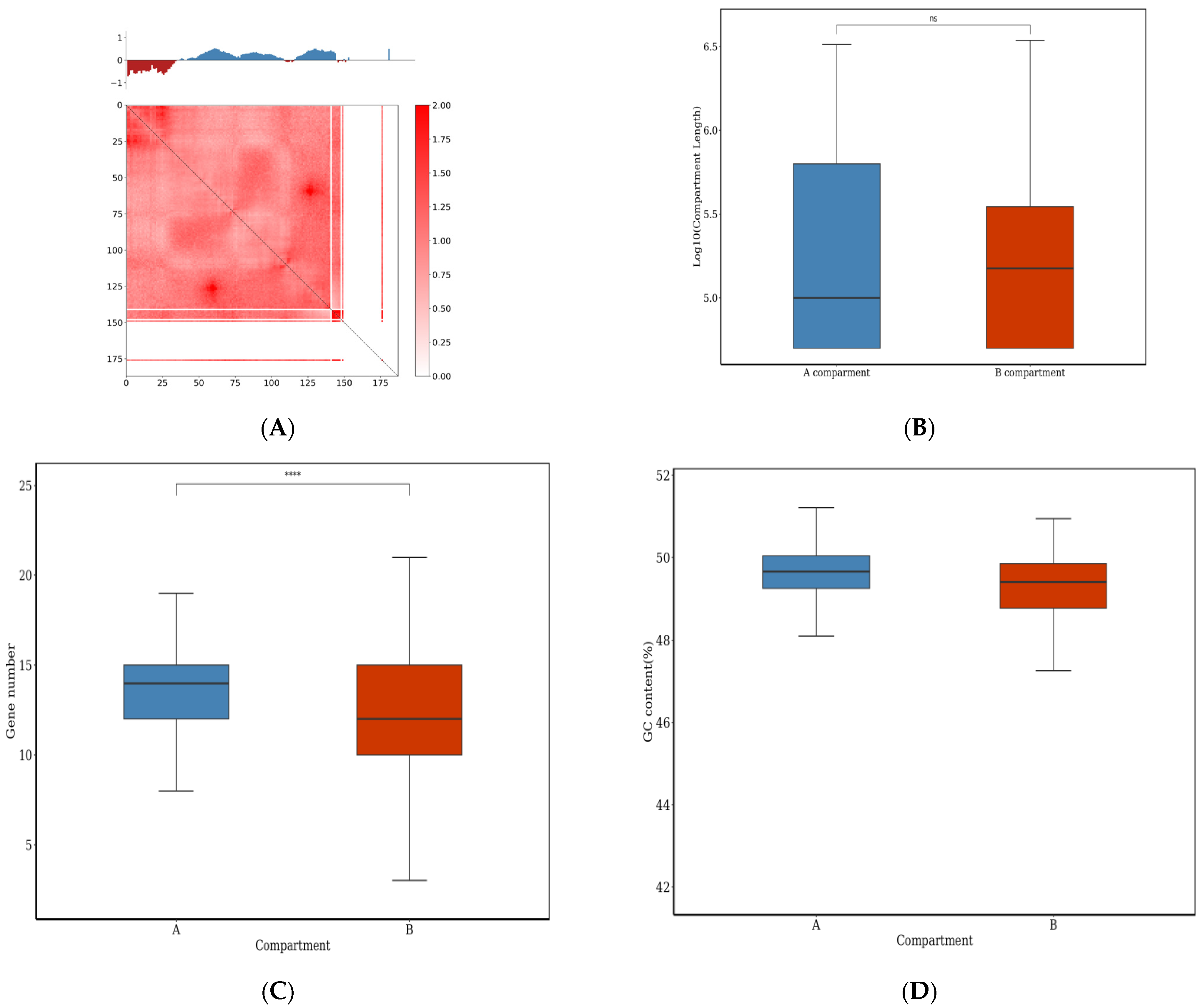
3.4. Three-Dimensional Genome Organization of E. tremulae
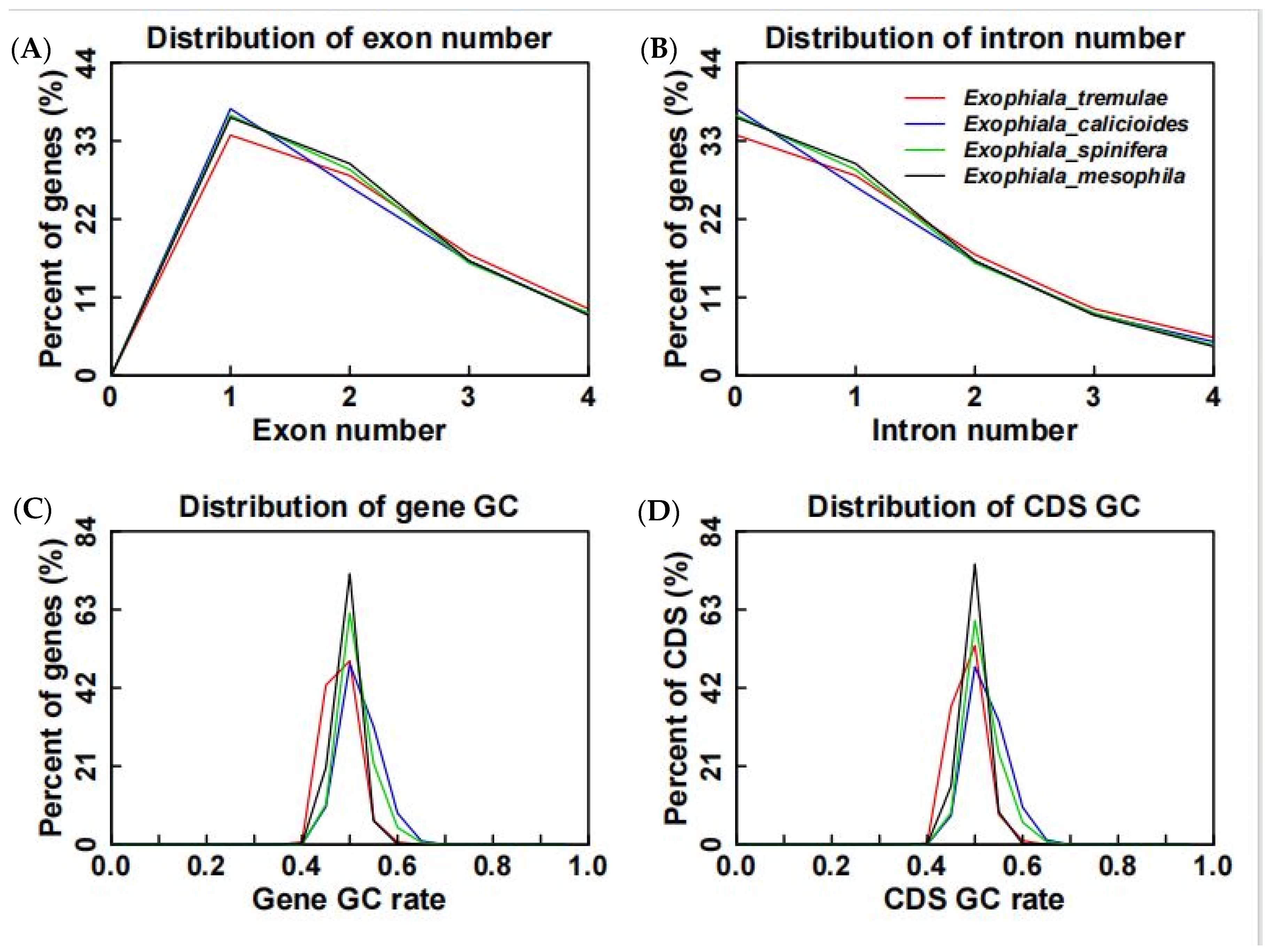
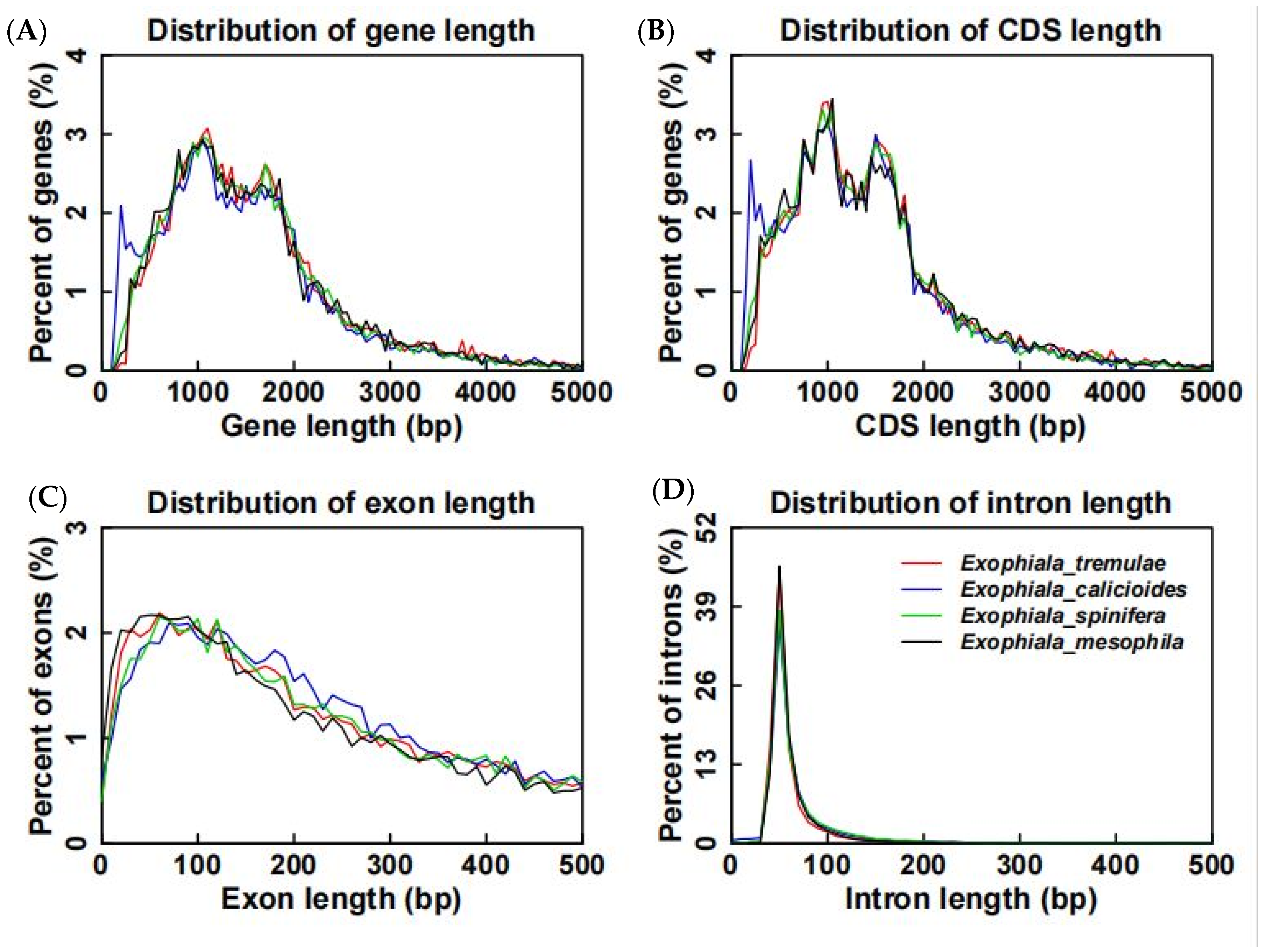
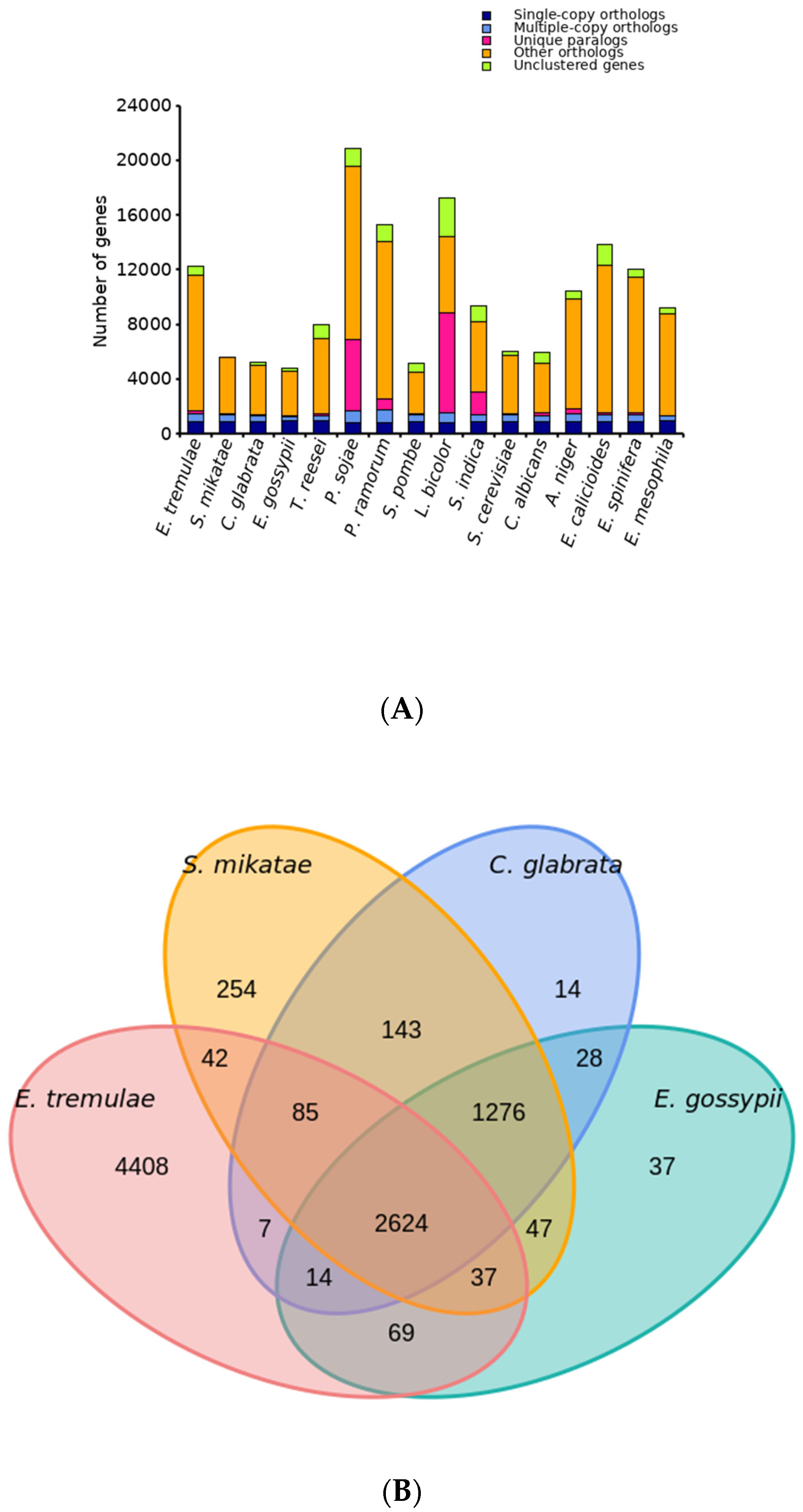
3.5. Comparative Analysis on Genomes of Different Fungi
4. Discussion
4.1. The Traits of the Genome of E. tremulae
4.2. Gene Family Expansion of the Genome of E. tremulae
4.3. Positive Selection of the E. tremulae Genome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santos, M.; Cesanelli, I.; Diánez, F.; Sánchez-Montesinos, B.; Moreno-Gavíra, A. Advances in the Role of Dark Septate Endophytes in the Plant Resistance to Abiotic and Biotic Stresses. J. Fungi 2021, 7, 939. [Google Scholar] [CrossRef]
- Jumpponen, A.; Trappe, J.M. Dark septate endophytes: A review of facultative biotrophic root-colonizing fungi. New Phytol. 1998, 140, 295–310. [Google Scholar] [CrossRef]
- Mandyam, K.; Jumpponen, A. Seeking the elusive function of the root-colonising dark septate endophytic fungi. Stud. Mycol. 2005, 53, 173–189. [Google Scholar] [CrossRef]
- Han, L.; Zuo, Y.; He, X.; Hou, Y.; Li, M.; Li, B. Plant identity and soil variables shift the colonisation and species composition of dark septate endophytes associated with medicinal plants in a northern farmland in China. Appl. Soil. Ecol. 2021, 167, 104042. [Google Scholar] [CrossRef]
- Qu, D.; Wu, F.; Guo, Y.; Zhang, J.; Li, M.; Yang, L.; Wang, L.; Su, H. Dark septate endophyte Anteaglonium sp. T010 promotes biomass accumulation in poplar by regulating sucrose metabolism and hormones. Tree Physiol. 2024, 44, tpae057. [Google Scholar] [CrossRef] [PubMed]
- Huertas, V.; Jiménez, A.; Diánez, F.; Chelhaoui, R.; Santos, M. Importance of Dark Septate Endophytes in Agriculture in the Face of Climate Change. J. Fungi 2024, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.Z.; Dai, M.D.; Zhu, J.N.; Liu, X.H.; Li, L.; Zhu, X.M.; Wang, J.Y.; Yuan, Z.L.; Lin, F.C. Dark septate endophyte Falciphora oryzae-assisted alleviation of cadmium in rice. J. Hazard. Mater. 2021, 419, 126435. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Z.; Zhang, G.; Liang, X.; Hu, L.; Li, Y.; He, Y.; Zhan, F. Dark septate endophyte Exophiala pisciphila promotes maize growth and alleviates cadmium toxicity. Front. Microbiol. 2023, 14, 1165131. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, G.; Liang, X.; Wang, L.; Li, Z.; He, Y.; Li, B.; Zhan, F. A Dark Septate Endophyte Improves Cadmium Tolerance of Maize by Modifying Root Morphology and Promoting Cadmium Binding to the Cell Wall and Phosphate. J. Fungi 2023, 9, 531. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Xu, M.; Ye, Q.; Gao, H.; He, X. Improved Tolerance of Artemisia ordosica to Drought Stress via Dark Septate Endophyte (DSE) Symbiosis. J. Fungi 2022, 8, 730. [Google Scholar] [CrossRef]
- Liu, N.; Jacquemyn, H.; Liu, Q.; Shao, S.-C.; Ding, G.; Xing, X. Effects of a Dark Septate Fungal Endophyte on the Growth and Physiological Response of Seedlings to Drought in an Epiphytic Orchid. Front. Microbiol. 2022, 13, 961172. [Google Scholar] [CrossRef]
- Farias, G.C.; Nunes, K.G.; Soares, M.A.; de Siqueira, K.A.; Lima, W.C.; Neves, A.L.R.; de Lacerda, C.F.; Filho, E.G. Dark septate endophytic fungi mitigate the effects of salt stress on cowpea plants. Braz. J. Microbiol. 2020, 51, 243–253. [Google Scholar] [CrossRef]
- Qu, D.; Wu, F.; Zhao, X.; Zhu, D.; Gu, L.; Yang, L.; Zhao, W.; Sun, Y.; Yang, J.; Tian, W.; et al. A bZIP transcription factor VabZIP12 from blueberry induced by dark septate endocyte improving the salt tolerance of transgenic Arabidopsis. Plant Sci. Int. J. Exp. Plant Biol. 2022, 315, 111135. [Google Scholar] [CrossRef]
- Gaber, D.A.; Berthelot, C.; Blaudez, D.; Kovács, G.M.; Franken, P. Impact of dark septate endophytes on salt stress alleviation of tomato plants. Front. Microbiol. 2023, 14, 1124879. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.Y.; Yue, Z.C.; Li, S.T.; Pan, Y.Y.; Chu, Z.Y.; Ban, Y.H.; Xu, Z.Y. Alleviation of Salt Stress and Changes in Glycyrrhizic Acid Accumulation by Dark Septate Endophytes in Glycyrrhiza glabra Grown under Salt Stress. J. Agric. Food Chem. 2024, 72, 14557–14569. [Google Scholar] [CrossRef]
- Haj Nuaima, R.; Ashrafi, S.; Maier, W.; Heuer, H. Fungi isolated from cysts of the beet cyst nematode parasitized its eggs and counterbalanced root damages. J. Pest. Sci. 2021, 94, 563–572. [Google Scholar] [CrossRef]
- Wang, K.; Wen, Z.; Asiegbu, F.O. The dark septate endophyte Phialocephala sphaeroides suppresses conifer pathogen transcripts and promotes root growth of Norway spruce. Tree Physiol. 2022, 42, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Terhonen, E.; Asiegbu, F.O. The dark septate endophyte Phialocephala sphaeroides confers growth fitness benefits and mitigates pathogenic effects of Heterobasidion on Norway spruce. Tree Physiol. 2022, 42, 891–906. [Google Scholar] [CrossRef]
- Luo, Q.; Hou, R.; Shang, X.; Li, S. Effects of Phosphorus-dissolving Dark Septate Endophytes on the Growth of Blueberry. J. Microbiol. 2023, 61, 837–851. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Liang, X.; Zhang, G.; Li, Z.; Yang, Z.; Zhan, F. The coexistence of arbuscular mycorrhizal fungi and dark septate endophytes synergistically enhanced the cadmium tolerance of maize. Front. Plant Sci. 2024, 15, 1349202. [Google Scholar] [CrossRef]
- Netherway, T.; Bengtsson, J.; Buegger, F.; Fritscher, J.; Oja, J.; Pritsch, K.; Hildebrand, F.; Krab, E.J.; Bahram, M. Pervasive associations between dark septate endophytic fungi with tree root and soil microbiomes across Europe. Nat. Commun. 2024, 15, 159. [Google Scholar] [CrossRef]
- Wu, M.H.; Xue, K.; Wei, P.J.; Jia, Y.L.; Zhang, Y.; Chen, S.Y. Soil microbial distribution and assembly are related to vegetation biomass in the alpine permafrost regions of the Qinghai-Tibet Plateau. Sci. Total Environ. 2022, 834, 155259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, H.; Lu, G.; Zhou, X.; Wang, J.; Li, J.; Zheng, K.; Fan, Y.; Zhou, H.; Wang, J.; et al. Non-targeted metabolomics analysis reveals the mechanism of arbuscular mycorrhizal symbiosis regulating the cold-resistance of Elymus nutans. Front. Microbiol. 2023, 14, 1134585. [Google Scholar] [CrossRef]
- Wei, X.; Sun, X.; Zhang, H.; Zhong, Q.; Lu, G. The influence of low-temperature resistant lactic acid bacteria on the enhancement of quality and the microbial community in winter Jerusalem Artichoke (Helianthus tuberosus L.) silage on the Qinghai-Tibet Plateau. Front. Microbiol. 2024, 15, 1297220. [Google Scholar] [CrossRef]
- Guo, J.; Xie, Z.; Meng, Q.; Xu, H.; Peng, Q.; Wang, B.; Dong, D.; Yang, J.; Jia, S. Distribution of rhizosphere fungi of Kobresia humilis on the Qinghai-Tibet Plateau. PeerJ 2024, 12, e16620. [Google Scholar] [CrossRef]
- Kumar, A.; Nandakumar, A.; Nair, S.; Singh, A.; Shashindran, N.; Thulasidharan, S.; Subhash, K.; Ramachandran, A.; Chowdhary, A. Exophiala dermatitidis as a cause of central line associated bloodstream infection in an infant: Case report and literature review. Rev. Iberoam. Micol. 2021, 38, 12–15. [Google Scholar] [CrossRef]
- Maraki, S.; Katzilakis, N.; Neonakis, I.; Stafylaki, D.; Meletiadis, J.; Hamilos, G.; Stiakaki, E. Exophiala dermatitidis Central Line-Associated Bloodstream Infection in a Child with Ewing’s Sarcoma: Case Report and Literature Review on Paediatric Infections. Mycopathologia 2022, 187, 595–602. [Google Scholar] [CrossRef]
- Mormeneo Bayo, S.; Bellés Bellés, A.; Prats Sánchez, I.; López González, E.; Aramburu Arnuelos, J.; Bernet Sánchez, A.; García González, M. Fungal corneal abscess caused by Exophiala dermatitidis. Rev. Esp. Quimioter. 2023, 36, 425–426. [Google Scholar] [CrossRef]
- Saraiva, M.; Beckmann, M.J.; Pflaum, S.; Pearson, M.; Carcajona, D.; Treasurer, J.W.; van West, P. Exophiala angulospora infection in hatchery-reared lumpfish (Cyclopterus lumpus) broodstock. J. Fish. Dis. 2019, 42, 335–343. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, S.; Li, S.; Wang, Y.; Li, L.; Xiao, B.; Li, J.; Long, L. Cerebral phaeohyphomycosis caused by Exophiala lecanii-corni. Travel. Med. Infect. Dis. 2023, 55, 102639. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Schumacher, R.K.; Akulov, A.; Bulgakov, T.S.; Carnegie, A.J.; Jurjević, Ž.; Decock, C.; Denman, S.; Lombard, L.; et al. New and Interesting Fungi. 3. Fungal Syst. Evol. 2020, 6, 157–231. [Google Scholar] [CrossRef] [PubMed]
- Crous, P.W.; Schubert, K.; Braun, U.; de Hoog, G.S.; Hocking, A.D.; Shin, H.D.; Groenewald, J.Z. Opportunistic, human-pathogenic species in the Herpotrichiellaceae are phenotypically similar to saprobic or phytopathogenic species in the Venturiaceae. Stud. Mycol. 2007, 58, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Crous, P.W.; Schumacher, R.K.; Wingfield, M.J.; Akulov, A.; Denman, S.; Roux, J.; Braun, U.; Burgess, T.I.; Carnegie, A.J.; Váczy, K.Z.; et al. New and Interesting Fungi. 1. Fungal Syst. Evol. 2018, 1, 169–216. [Google Scholar] [CrossRef]
- Tibpromma, S.; Hyde, K.D.; Jeewon, R.; Maharachchikumbura, S.S.N.; Liu, J.-K.; Bhat, D.J.; Jones, E.B.G.; McKenzie, E.H.C.; Camporesi, E.; Bulgakov, T.S.; et al. Fungal diversity notes 491–602: Taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers. 2017, 83, 1–261. [Google Scholar] [CrossRef]
- Crous, P.W.; Cowan, D.A.; Maggs-Kölling, G.; Yilmaz, N.; Larsson, E.; Angelini, C.; Brandrud, T.E.; Dearnaley, J.D.W.; Dima, B.; Dovana, F.; et al. Fungal Planet description sheets: 1112-1181. Persoonia 2020, 45, 251–409. [Google Scholar] [CrossRef]
- Xiao, Y.; Dai, M.-X.; Zhang, G.-Q.; Yang, Z.-X.; He, Y.-M.; Zhan, F.-D. Effects of the Dark Septate Endophyte (DSE) Exophiala pisciphila on the Growth of Root Cell Wall Polysaccharides and the Cadmium Content of Zea mays L. under Cadmium Stress. J. Fungi 2021, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.L.; Hamayun, M.; Ahmad, N.; Waqas, M.; Kang, S.-M.; Kim, Y.-H.; Lee, I.-J. Exophiala sp. LHL08 reprograms Cucumis sativus to higher growth under abiotic stresses. Physiol. Plant 2011, 143, 329–343. [Google Scholar] [CrossRef]
- Maciá-Vicente, J.G.; Glynou, K.; Piepenbring, M. A new species of Exophiala associated with roots. Mycol. Progress. 2016, 15, 18. [Google Scholar] [CrossRef]
- Crous, P.W.; Cowan, D.A.; Maggs-Kölling, G.; Yilmaz, N.; Thangavel, R.; Wingfield, M.J.; Noordeloos, M.E.; Dima, B.; Brandrud, T.E.; Jansen, G.M.; et al. Fungal Planet description sheets: 1182–1283. Persoonia 2021, 46, 313–528. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Shivas, R.G.; Edwards, J.; Seifert, K.A.; Alfenas, A.C.; Alfenas, R.F.; Burgess, T.I.; Carnegie, A.J.; Hardy, G.E.S.J.; et al. Fungal Planet description sheets: 69-91. Persoonia 2011, 26, 108–156. [Google Scholar] [CrossRef]
- Yu, Y.; Teng, Z.; Mou, Z.; Lv, Y.; Li, T.; Chen, S.; Zhao, D.; Zhao, Z. Melatonin confers heavy metal-induced tolerance by alleviating oxidative stress and reducing the heavy metal accumulation in Exophiala pisciphila, a dark septate endophyte (DSE). BMC Microbiol. 2021, 21, 40. [Google Scholar] [CrossRef]
- Domb, K.; Wang, N.; Hummel, G.; Liu, C. Spatial Features and Functional Implications of Plant 3D Genome Organization. Annu. Rev. Plant Biol. 2022, 73, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Huang, L.; Huang, J.; Zhang, H.; Huang, Y.; Benhamed, M.; Wang, M.; Chen, X.; Zhang, M.; Liu, T.; et al. Folding Features and Dynamics of 3D Genome Architecture in Plant Fungal Pathogens. Microbiol. Spectr. 2022, 10, e0260822. [Google Scholar] [CrossRef]
- Ni, L.; Liu, Y.; Ma, X.; Liu, T.; Yang, X.; Wang, Z.; Liang, Q.; Liu, S.; Zhang, M.; Wang, Z.; et al. Pan-3D genome analysis reveals structural and functional differentiation of soybean genomes. Genome Biol. 2023, 24, 12. [Google Scholar] [CrossRef]
- Tourdot, E.; Grob, S. Three-dimensional chromatin architecture in plants—General features and novelties. Eur. J. Cell Biol. 2023, 102, 151344. [Google Scholar] [CrossRef]
- Li, C.-X.; Liu, L.; Zhang, T.; Luo, X.-M.; Feng, J.-X.; Zhao, S. Three-Dimensional Genome Map of the Filamentous Fungus Penicillium oxalicum. Microbiol. Spectr. 2022, 10, e02121. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, L.; Chen, H.; Ye, W.; Dong, S.; Zheng, X.; Wang, Y. ATAC-Seq Reveals the Landscape of Open Chromatin and cis-Regulatory Elements in the Phytophthora sojae Genome. Mol. Plant Microbe Interact. 2022, 35, 301–310. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from plant tissues. In Plant Molecular Biology Manual; Gelvin, S.B., Schilperoort, R.A., Verma, D.P.S., Eds.; Springer: Dordrecht, The Netherlands, 1989; pp. 73–83. [Google Scholar]
- Padmarasu, S.; Himmelbach, A.; Mascher, M.; Stein, N. In Situ Hi-C for Plants: An Improved Method to Detect Long-Range Chromatin Interactions. Methods Mol. Biol. 2019, 1933, 441–472. [Google Scholar] [CrossRef]
- Cheng, H.; Jarvis, E.D.; Fedrigo, O.; Koepfli, K.-P.; Urban, L.; Gemmell, N.J.; Li, H. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 2022, 40, 1332–1335. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 4, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Jurka, J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000, 16, 418–420. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Gertz, E.M.; Yu, Y.-K.; Agarwala, R.; Schäffer, A.A.; Altschul, S.F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 2006, 4, 41. [Google Scholar]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [PubMed]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez Alvarado, A.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Han, M.V.; Thomas, G.W.C.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Hansen, K.D. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015, 16, 180. [Google Scholar] [CrossRef]
- Mouchiroud, D.; D’Onofrio, G.; Aïssani, B.; Macaya, G.; Gautier, C.; Bernardi, G. The distribution of genes in the human genome. Gene 1991, 100, 181–187. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, B.R.; Dekker, J.; Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods (San Diego Calif.) 2015, 72, 65–75. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Armstrong, M.R.; Whisson, S.C.; Pritchard, L.; Bos, J.I.B.; Venter, E.; Avrova, A.O.; Rehmany, A.P.; Böhme, U.; Brooks, K.; Cherevach, I.; et al. An ancestral oomycete locus contains late blight avirulence gene Avr3a, encoding a protein that is recognized in the host cytoplasm. Proc. Natl. Acad. Sci. USA 2005, 102, 7766–7771. [Google Scholar] [CrossRef] [PubMed]
- Rehmany, A.P.; Gordon, A.; Rose, L.E.; Allen, R.L.; Armstrong, M.R.; Whisson, S.C.; Kamoun, S.; Tyler, B.M.; Birch, P.R.J.; Beynon, J.L. Differential recognition of highly divergent downy mildew avirulence gene alleles by RPP1 resistance genes from two Arabidopsis lines. Plant Cell 2005, 17, 1839–1850. [Google Scholar] [CrossRef]
- Jiang, R.H.Y.; Tripathy, S.; Govers, F.; Tyler, B.M. RXLR effector reservoir in two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc. Natl. Acad. Sci. USA 2008, 105, 4874–4879. [Google Scholar] [CrossRef]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef]
- Baxter, L.; Tripathy, S.; Ishaque, N.; Boot, N.; Cabral, A.; Kemen, E.; Thines, M.; Ah-Fong, A.; Anderson, R.; Badejoko, W.; et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 2010, 330, 1549–1551. [Google Scholar] [CrossRef]
- Yin, J.; Gu, B.; Huang, G.; Tian, Y.; Quan, J.; Lindqvist-Kreuze, H.; Shan, W. Conserved RXLR Effector Genes of Phytophthora infestans Expressed at the Early Stage of Potato Infection Are Suppressive to Host Defense. Front. Plant Sci. 2017, 8, 2155. [Google Scholar] [CrossRef]
- Zuccaro, A.; Lahrmann, U.; Güldener, U.; Langen, G.; Pfiffi, S.; Biedenkopf, D.; Wong, P.; Samans, B.; Grimm, C.; Basiewicz, M.; et al. Endophytic life strategies decoded by genome and transcriptome analyses of the mutualistic root symbiont Piriformospora indica. PLoS Pathog. 2011, 7, e1002290. [Google Scholar] [CrossRef]
- Liu, L.; Xu, L.; Jia, Q.; Pan, R.; Oelmüller, R.; Zhang, W.; Wu, C. Arms race: Diverse effector proteins with conserved motifs. Plant Signal. Behav. 2019, 14, 1557008. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; McLellan, H.; Boevink, P.C.; Birch, P.R.J. RxLR Effectors: Master Modulators, Modifiers and Manipulators. Mol. Plant-Microbe Interact. MPMI 2023, 36, 754–763. [Google Scholar] [CrossRef]
- Qiu, X.; Kong, L.; Chen, H.; Lin, Y.; Tu, S.; Wang, L.; Chen, Z.; Zeng, M.; Xiao, J.; Yuan, P.; et al. The Phytophthora sojae nuclear effector PsAvh110 targets a host transcriptional complex to modulate plant immunity. Plant Cell 2023, 35, 574–597. [Google Scholar] [CrossRef]
- Gao, H.; Guo, Y.; Ren, M.; Tang, L.; Gao, W.; Tian, S.; Shao, G.; Peng, Q.; Gu, B.; Miao, J.; et al. Phytophthora RxLR effector PcSnel4B promotes degradation of resistance protein AtRPS2. Plant Physiol. 2023, 193, 1547–1560. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.; Huang, Y.; Zheng, J.; Yang, Y.; Cao, Y.; Ding, L.; Meng, Y.; Shan, W. Phytophthora infestans RXLR effector Pi23014 targets host RNA-binding protein NbRBP3a to suppress plant immunity. Mol. Plant Pathol. 2024, 25, e13416. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.; Kost, G.W.; Oelmüller, R. Piriformospora Indica: Sebacinales and Their Biotechnologicala Pplications; Springer: Berlin, Germany, 2013. [Google Scholar] [CrossRef]
- Gill, S.S.; Gill, R.; Trivedi, D.K.; Anjum, N.A.; Sharma, K.K.; Ansari, M.W.; Ansari, A.A.; Johri, A.K.; Prasad, R.; Pereira, E.; et al. Piriformospora indica: Potential and Significance in Plant Stress Tolerance. Front. Microbiol. 2016, 7, 332. [Google Scholar] [CrossRef]
- Das, A.; Kamal, S.; Shakil, N.A.; Sherameti, I.; Oelmüller, R.; Dua, M.; Tuteja, N.; Johri, A.K.; Varma, A. The root endophyte fungus Piriformospora indica leads to early flowering, higher biomass and altered secondary metabolites of the medicinal plant, Coleus forskohlii. Plant Signal. Behav. 2012, 7, 103–112. [Google Scholar] [CrossRef]
- Kim, D.; Abdelaziz, M.E.; Ntui, V.O.; Guo, X.; Al-Babili, S. Colonization by the endophyte Piriformospora indica leads to early flowering in Arabidopsis thaliana likely by triggering gibberellin biosynthesis. Biochem. Biophys. Res. Commun. 2017, 490, 1162–1167. [Google Scholar] [CrossRef]
- Pan, R.; Xu, L.; Wei, Q.; Wu, C.; Tang, W.; Oelmüller, R.; Zhang, W. Piriformospora indica promotes early flowering in Arabidopsis through regulation of the photoperiod and gibberellin pathways. PLoS ONE 2017, 12, e0189791. [Google Scholar] [CrossRef]
- Benini, S. Carbohydrate-Active Enzymes: Structure, Activity, and Reaction Products. Int. J. Mol. Sci. 2020, 21, 2727. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.W.; Im, J.H.; Kong, W.S.; Park, Y.J. Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome. Microorganisms 2020, 9, 20. [Google Scholar] [CrossRef]
- Knapp, D.G.; Németh, J.B.; Barry, K.; Hainaut, M.; Henrissat, B.; Johnson, J.; Kuo, A.; Lim, J.H.P.; Lipzen, A.; Nolan, M.; et al. Comparative genomics provides insights into the lifestyle and reveals functional heterogeneity of dark septate endophytic fungi. Sci. Rep. 2018, 8, 6321. [Google Scholar] [CrossRef]
- Kumar, K.; Singal, S.; Goyal, A. Role of carbohydrate binding module (CBM3c) of GH9 β-1,4 endoglucanase (Cel9W) from Hungateiclostridium thermocellum ATCC 27405 in catalysis. Carbohydr Res 2019, 484, 107782. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Zhang, D.; Jiang, W.; Zhang, H.; Cheng, W.; Li, H.; Liu, J.; Jiang, Z. How carbohydrate-binding module affects the catalytic properties of endoglucanase. Int. J. Biol. Macromol. 2024, 278, 134653. [Google Scholar] [CrossRef]
- Fabri, J.H.T.M.; Rocha, M.C.; Fernandes, C.M.; Campanella, J.E.M.; Cunha, A.F.d.; Del Poeta, M.; Malavazi, I. The Heat Shock Transcription Factor HsfA Plays a Role in Membrane Lipids Biosynthesis Connecting Thermotolerance and Unsaturated Fatty Acid Metabolism in Aspergillus fumigatus. Microbiol. Spectr. 2023, 11, e0162723. [Google Scholar] [CrossRef]
- Li, Y.; Yan, P.; Lei, Q.; Li, B.; Sun, Y.; Li, S.; Lei, H.; Xie, N. Metabolic adaptability shifts of cell membrane fatty acids of Komagataeibacter hansenii HDM1-3 improve acid stress resistance and survival in acidic environments. J. Ind. Microbiol. Biotechnol. 2019, 46, 1491–1503. [Google Scholar] [CrossRef]
- Mauger, M.; Ferreri, C.; Chatgilialoglu, C.; Seemann, M. The bacterial protective armor against stress: The cis-trans isomerase of unsaturated fatty acids, a cytochrome-c type enzyme. J. Inorg. Biochem. 2021, 224, 111564. [Google Scholar] [CrossRef]
- Loffeld, B.; Keweloh, H. cis/trans isomerization of unsaturated fatty acids as possible control mechanism of membrane fluidity in Pseudomonas putida P8. Lipids 1996, 31, 811–815. [Google Scholar]
- Eberlein, C.; Baumgarten, T.; Starke, S.; Heipieper, H.J. Immediate response mechanisms of Gram-negative solvent-tolerant bacteria to cope with environmental stress: Cis-trans isomerization of unsaturated fatty acids and outer membrane vesicle secretion. Appl. Microbiol. Biotechnol. 2018, 102, 2583–2593. [Google Scholar] [CrossRef]
- Zhang, H.W.; Lin, Z.X.; Tung, Y.S.; Kwan, T.H.; Mok, C.K.; Leung, C.; Chan, L.S. Cordyceps sinensis (a traditional Chinese medicine) for treating chronic kidney disease. Cochrane Database Syst. Rev. 2014, 2014, Cd008353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Chen, Y.-H.; Pan, B.-S.; Chang, M.-M.; Huang, B.-M. Functional study of Cordyceps sinensis and cordycepin in male reproduction: A review. J. Food Drug Anal. 2017, 25, 197–205. [Google Scholar] [CrossRef]
- Steyer, J.T.; Todd, R.B. Branched-chain amino acid biosynthesis in fungi. Essays Biochem. 2023, 67, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Schertl, P.; Danne, L.; Braun, H.P. 3-Hydroxyisobutyrate Dehydrogenase Is Involved in Both, Valine and Isoleucine Degradation in Arabidopsis thaliana. Plant Physiol. 2017, 175, 51–61. [Google Scholar] [CrossRef]
- Blank, H.M.; Reuse, C.; Schmidt-Hohagen, K.; Hammer, S.E.; Hiller, K.; Polymenis, M. Branched-chain amino acid synthesis is coupled to TOR activation early in the cell cycle in yeast. EMBO Rep. 2023, 24, e57372. [Google Scholar] [CrossRef] [PubMed]
- Mostofian, B.; Zhuang, T.; Cheng, X.; Nickels, J.D. Branched-Chain Fatty Acid Content Modulates Structure, Fluidity, and Phase in Model Microbial Cell Membranes. J. Phys. Chem. B 2019, 123, 5814–5821. [Google Scholar] [CrossRef]
- Chen, X.; Teoh, W.P.; Stock, M.R.; Resko, Z.J.; Alonzo, F. Branched chain fatty acid synthesis drives tissue-specific innate immune response and infection dynamics of Staphylococcus aureus. PLoS Pathog. 2021, 17, e1009930. [Google Scholar] [CrossRef]
- Davis, I.; Yang, Y.; Wherritt, D.; Liu, A. Reassignment of the human aldehyde dehydrogenase ALDH8A1 (ALDH12) to the kynurenine pathway in tryptophan catabolism. J. Biol. Chem. 2018, 293, 9594–9603. [Google Scholar] [CrossRef]
- Kang, Z.; Zhang, J.; Zhou, J.; Qi, Q.; Du, G.; Chen, J. Recent advances in microbial production of δ-aminolevulinic acid and vitamin B12. Biotechnol. Adv. 2012, 30, 1533–1542. [Google Scholar] [CrossRef]
- Lake, M.W.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature 2001, 414, 325–329. [Google Scholar]
- Lois, L.M.; Lima, C.D. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005, 24, 439–451. [Google Scholar] [PubMed]
- Lee, I.; Schindelin, H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 2008, 134, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Yeh, E.T.H. SUMO: From Bench to Bedside. Physiol. Rev. 2020, 100, 1599–1619. [Google Scholar] [CrossRef] [PubMed]
- Zakelj-Mavric, M.; Kastelic-Suhadolc, T.; Plemenitas, A.; Rizner, T.L.; Belic, I. Steroid hormone signalling system and fungi. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1995, 112, 637–642. [Google Scholar] [CrossRef]
- Jiang, Y.-Q.; Lin, J.-P. Recent progress in strategies for steroid production in yeasts. World J. Microbiol. Biotechnol. 2022, 38, 93. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Test |
|---|---|
| Number of contigs | 29 |
| Assembly length (bp) | 51,903,848 |
| Contig N50 (bp) | 4,428,737 |
| anchor ratio (%) | 99.331 |
| HiFi reads mapping rate (%) | 99.58 |
| HiFi reads coverage (%) | 99.96 |
| Complete BUSCOs of genome (%) | 99.8 |
| QV | 64.7991 |
| Term | Genes | Percentage (%) |
|---|---|---|
| Complete BUSCOs of genome | 757 | 99.8 |
| Complete and single-copy BUSCOs | 753 | 99.3 |
| Complete and duplicated BUSCOs | 4 | 0.5 |
| Fragmented BUSCOs | 1 | 0.1 |
| Missing BUSCOs | 0 | 0 |
| Total BUSCO groups searched | 758 | 100 |
| Number | Percent (%) | |
| Total | 12,277 | |
| InterPro | 9362 | 76.26 |
| GO | 9307 | 75.81 |
| KEGG_ALL | 11,324 | 92.24 |
| KEGG_KO | 4004 | 32.61 |
| Swissprot | 8003 | 65.19 |
| TrEMBL | 11,917 | 97.07 |
| NR | 11,918 | 97.08 |
| Annotated | 11,932 | 97.19 |
| Unannotated | 345 | 2.81 |
| Species | GHs | GTs | PLs | CEs | CBMs | AAs | Total |
|---|---|---|---|---|---|---|---|
| Fungi in the genus Exophiala | |||||||
| Exophiala tremulae CICC2537 | 170 | 102 | 1 | 16 | 5 | 82 | 376 |
| Exophiala dermatitidis UT8656 | 123 | 92 | 2 | 5 | 8 | 36 | 266 |
| Exophiala spinifera CBS89968 | 153 | 114 | 1 | 8 | 11 | 42 | 329 |
| Exophiala mesophila CBS40295 | 121 | 112 | 1 | 5 | 11 | 48 | 298 |
| Exophiala xenobiotica CBS118157 | 189 | 119 | 2 | 9 | 14 | 68 | 401 |
| Exophiala oligosperma CBS72588 | 168 | 137 | 0 | 10 | 15 | 65 | 395 |
| Exophiala aquamarina CBS119918 | 193 | 113 | 1 | 8 | 12 | 52 | 379 |
| Exophiala sideris CBS121828 | 146 | 100 | 0 | 6 | 14 | 46 | 312 |
| Root endophytic fungi | |||||||
| Serendipita indica DSM 11827 | 176 | 72 | 14 | 40 | 61 | 56 | 419 |
| Trichoderma reesei QM6a | 198 | 97 | 6 | 10 | 25 | 31 | 367 |
| Trichoderma harzianum TR274 | 171 | 101 | 8 | 19 | 38 | 38 | 385 |
| Arbuscular mycorrhizal fungi | |||||||
| Rhizophagus irregularis A1 (RhiirA1_1) | 32 | 105 | 9 | 10 | 17 | 34 | 207 |
| Gigaspora rosea v1.0 (Gigro1) | 102 | 196 | 3 | 23 | 16 | 60 | 400 |
| Ectomycorrhizal fungi | |||||||
| Cenococcum geophilum 1.58 (Cenge3) | 171 | 93 | 2 | 11 | 30 | 50 | 357 |
| Lactarius deliciosus 48 v1.0 (Lacdel1) | 156 | 104 | 8 | 12 | 36 | 64 | 380 |
| Laccaria bicolor v2.0 (Lacbi2) | 185 | 97 | 9 | 18 | 29 | 61 | 399 |
| Types of Effector Proteins | Numbers |
|---|---|
| Total predicted effector proteins | 3337 |
| Apoplastic effector | 163 |
| Apoplastic/cytoplasmic effector | 26 |
| Cytoplasmic effector | 3100 |
| Cytoplasmic/apoplastic effector | 48 |
| CRN | 172 |
| LXAR | 605 |
| RXLR | 1112 |
| CFEM | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Fan, J.; Hu, D.; Sun, H.; Lu, G.; Wang, Y.; Yang, Y. The Three-Dimensional Structure of the Genome of the Dark Septate Endophyte Exophiala tremulae and Its Symbiosis Effect on Alpine Meadow Plant Growth. J. Fungi 2025, 11, 246. https://doi.org/10.3390/jof11040246
Wu C, Fan J, Hu D, Sun H, Lu G, Wang Y, Yang Y. The Three-Dimensional Structure of the Genome of the Dark Septate Endophyte Exophiala tremulae and Its Symbiosis Effect on Alpine Meadow Plant Growth. Journal of Fungi. 2025; 11(4):246. https://doi.org/10.3390/jof11040246
Chicago/Turabian StyleWu, Chu, Junjie Fan, Die Hu, Honggang Sun, Guangxin Lu, Yun Wang, and Yujie Yang. 2025. "The Three-Dimensional Structure of the Genome of the Dark Septate Endophyte Exophiala tremulae and Its Symbiosis Effect on Alpine Meadow Plant Growth" Journal of Fungi 11, no. 4: 246. https://doi.org/10.3390/jof11040246
APA StyleWu, C., Fan, J., Hu, D., Sun, H., Lu, G., Wang, Y., & Yang, Y. (2025). The Three-Dimensional Structure of the Genome of the Dark Septate Endophyte Exophiala tremulae and Its Symbiosis Effect on Alpine Meadow Plant Growth. Journal of Fungi, 11(4), 246. https://doi.org/10.3390/jof11040246