
Three-Dimensional Structural Heteromorphs of Mating-Type Proteins in Hirsutella sinensis and the Natural Cordyceps sinensis Insect–Fungal Complex


Abstract
1. Introduction
2. Materials and Methods
2.1. C. sinensis Isolates and Accession Numbers of the MAT1-1-1 and MAT1-2-1 Proteins
2.2. Genome, Transcriptome, and Metatranscriptome Assemblies of H. sinensis Strains and the Natural C. sinensis Insect–Fungal Complex
2.3. Statistical Clustering Analysis for the MAT1-1-1 and MAT1-2-1 Protein Sequences
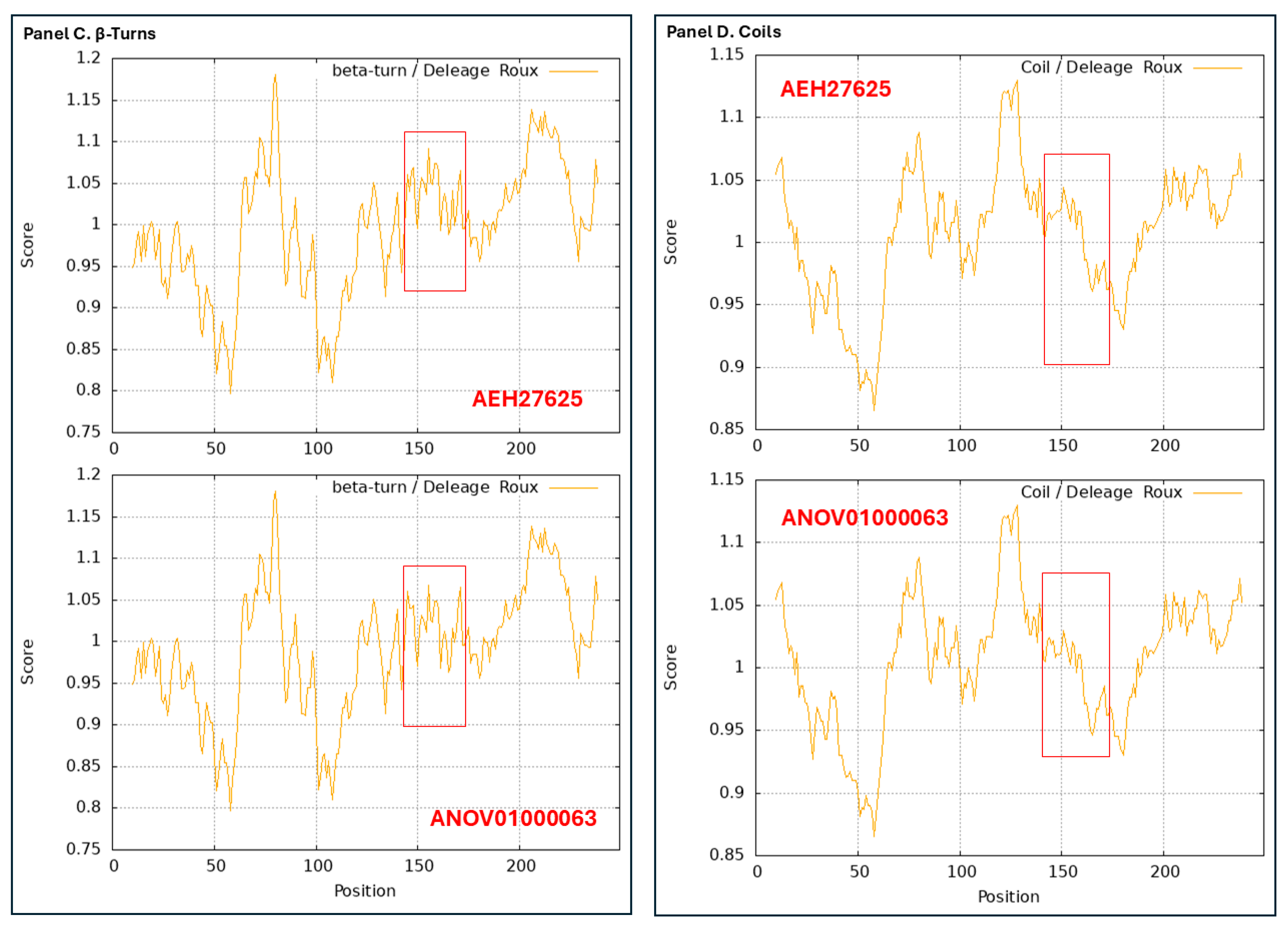
2.4. AlphaFold-Based Prediction of 3D Structures of Mating Proteins
 very high confidence (pLDDT > 90) residues are shown in dark blue,
very high confidence (pLDDT > 90) residues are shown in dark blue,  high (90 > pLDDT > 70) in light blue,
high (90 > pLDDT > 70) in light blue,  low (70 > pLDDT > 50) in yellow, and
low (70 > pLDDT > 50) in yellow, and  very low (pLDDT < 50) in orange [Mariani et al., 2013 [55]; Wroblewski & Kmiecik 2024 [58]]. Note that a protein region that is assigned a low pLDDT score does not necessarily indicate that this region is the most variable region in the protein sequence; in contrast, a substantially variable region of a protein may be assigned a high pLDDT score. The AlphaFold database provides an average pLDDT score for each of the predicted 3D structure models of mating proteins, representing the overall model confidence in the predicted 3D structures.
very low (pLDDT < 50) in orange [Mariani et al., 2013 [55]; Wroblewski & Kmiecik 2024 [58]]. Note that a protein region that is assigned a low pLDDT score does not necessarily indicate that this region is the most variable region in the protein sequence; in contrast, a substantially variable region of a protein may be assigned a high pLDDT score. The AlphaFold database provides an average pLDDT score for each of the predicted 3D structure models of mating proteins, representing the overall model confidence in the predicted 3D structures.2.5. Alignment Analysis of Protein Sequences
2.6. Amino Acid Properties and Scale Analysis
3. Results
3.1. Diversity of the MAT1-1-1 and MAT1-2-1 Proteins in H. sinensis Strains and Wild-Type C. sinensis Isolates on the Basis of the AlphaFold-Predicted 3D Structures
3.2. Bayesian Analysis of the MAT1-1-1 and MAT1-2-1 Proteins
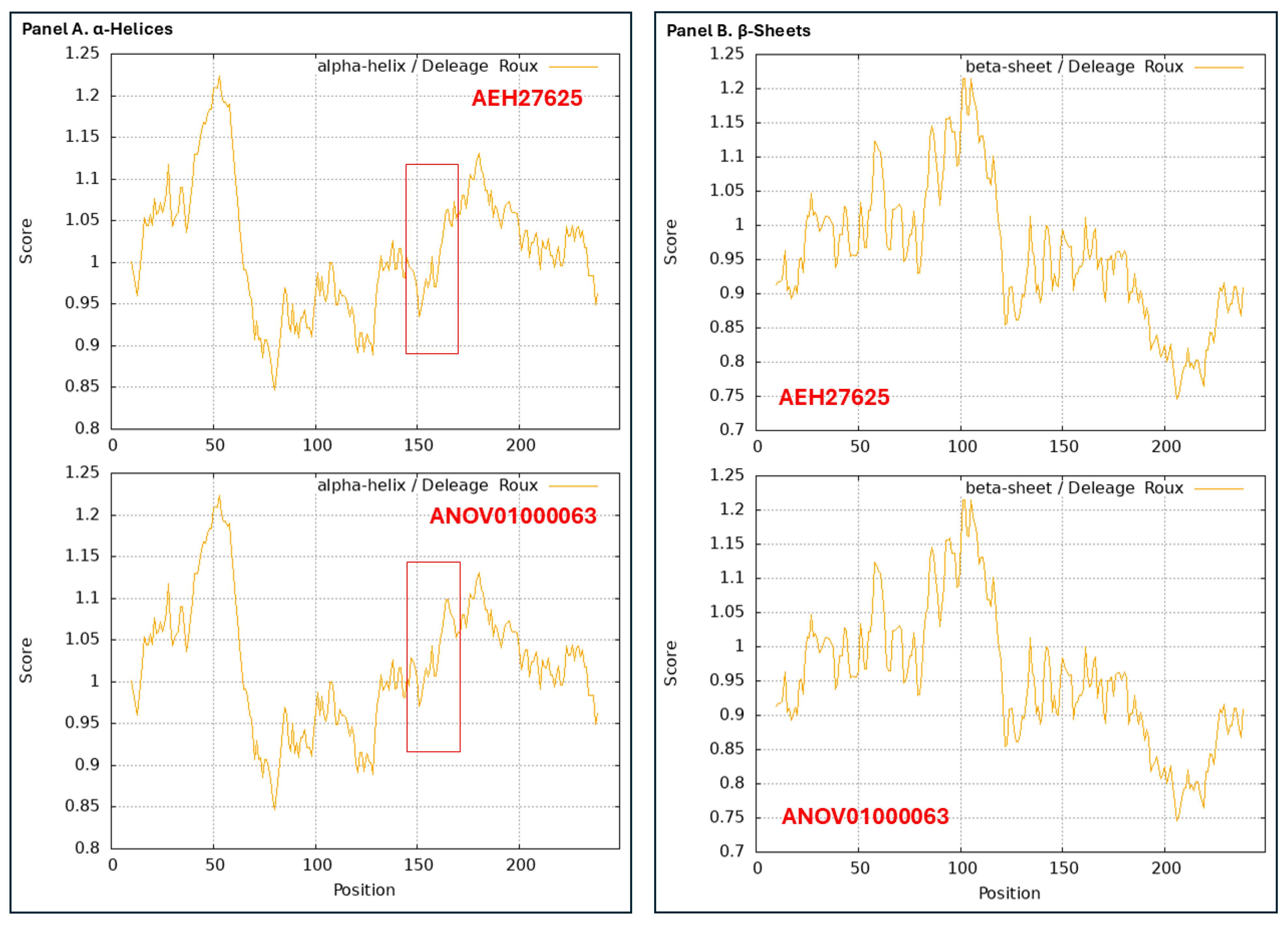
3.3. Heteromorphic AlphaFold-Predicted 3D Structures of the MAT1-1-1 Proteins
3.4. Heteromorphic AlphaFold-Predicted 3D Structures of the MAT1-2-1 Proteins
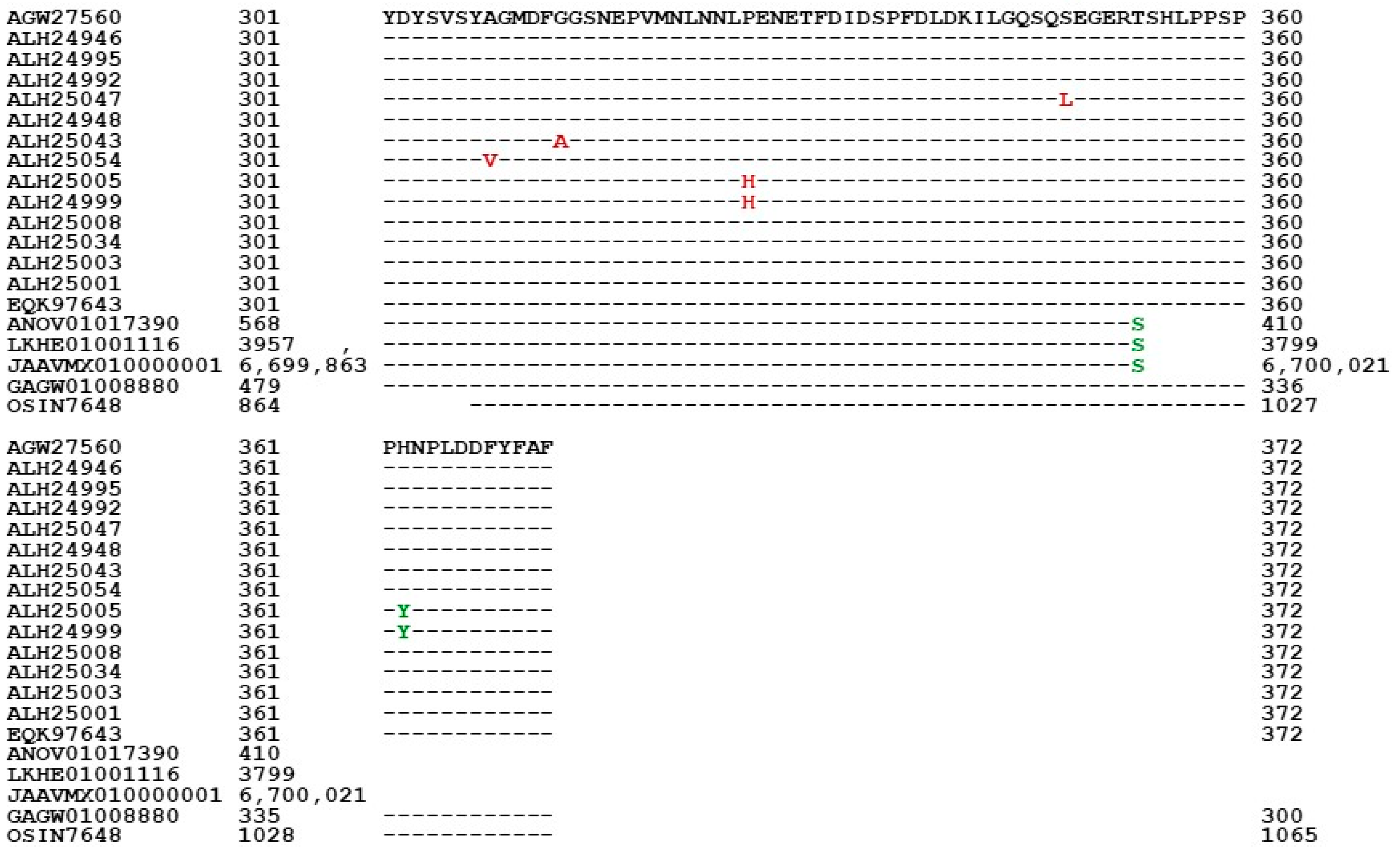
3.5. Primary Structures of the MAT1-1-1 Proteins
3.6. Primary Structures of the MAT1-2-1 Proteins
3.7. Differential Genomic Occurrence of the MAT1-1-1 and MAT1-2-1 Proteins in H. sinensis
3.8. Differential Transcriptomic and Metatranscriptomic Occurrences of the MAT1-1-1 and MAT1-2-1 Proteins in H. sinensis and the Natural C. sinensis Insect–Fungal Complex
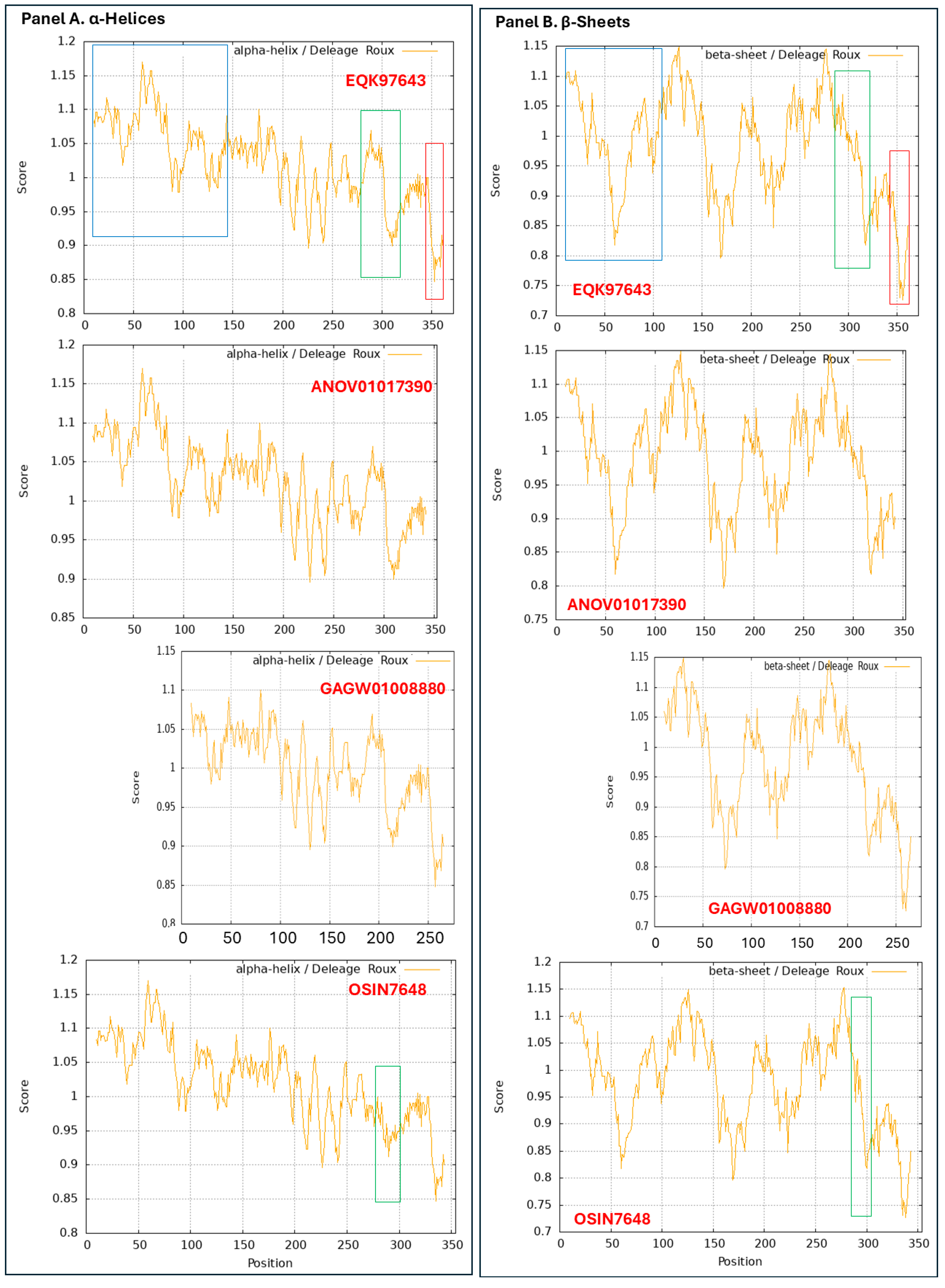
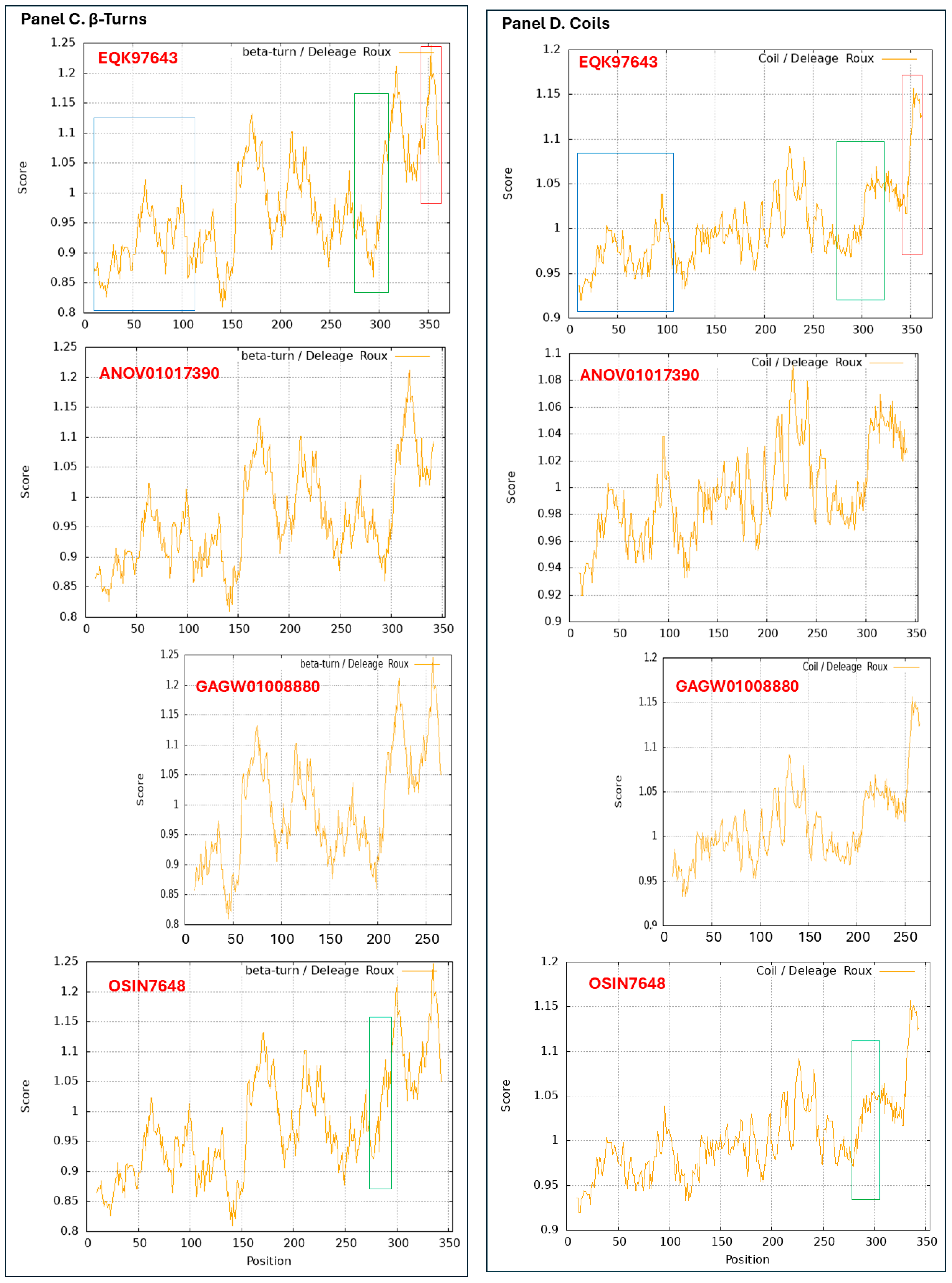
3.9. Diverse Secondary (2D) Structures of the MAT1-1-1 Proteins Encoded by the Genome of H. sinensis and the Metatranscriptome of Natural C. sinensis Insect–Fungal Complex
3.10. Diverse 2D Structures of the MAT1-2-1 Proteins Encoded by the Genomes, Transcriptomes, and Metatranscriptomes of H. sinensis and the Natural C. sinensis Insect–Fungal Complex
4. Discussion
4.1. Protein 3D Structure Analysis via the AI-Based AlphaFold Prediction System in Combination with Statistical Bayesian Clustering Technology to Stratify 3D Structure Models
4.2. Heteromorphic 3D Structures of the MAT1-1-1 and MAT1-2-1 Proteins in H. sinensis Strains and Wild-Type C. sinensis Isolates
4.3. Differential Occurrences of MAT1-1-1 and MAT1-2-1 Proteins with Heteromorphic Sereostructures in H. sinensis Strains and C. sinensis Isolates
4.4. Heteromorphic 3D Structures of the Mating Proteins and Sexual Reproductive Behavior of H. sinensis, Genotype #1 of O. sinensis
4.5. Heteromorphic 3D Structures of the Mating Proteins and Sexual Reproduction Strategy During the Lifecycle of the Natural C. sinensis Insect–Fungi Complex
- (1)
- Evidence for the differential cooccurrence of multiple genotypes of O. sinensis in the compartments of the natural C. sinensis insect–fungi complex is as follows:
- (1-a).
- Differential occurrence of AT-biased Genotype #4 or #5 of O. sinensis without the cooccurrence of GC-biased H. sinensis in natural C. sinensis samples collected from different production areas in geographically remote locations [Engh 1999 [90]; Kinjo and Zang 2001 [68]; Stensrud et al., 2005 [91], 2007 [92]; Mao et al., 2013 [71]];
- (1-b).
- Multiple cooccurring GC- and AT-biased genotypes of O. sinensis have been observed differentially in different combinations in the stroma, caterpillar body, ascocarps, and ascospores of natural C. sinensis [Xiao et al., 2009 [83]; Zhu et al., 2010 [84]; Li et al., 2013 [70], 2022 [6], 2023c [49], 2023d [15]; Mao et al., 2013 [71]]. The abundances of the O. sinensis genotypes underwent dynamic alterations in an asynchronous, disproportional manner in the caterpillar bodies and stromata of C. sinensis specimens during maturation, with a consistent predominance of the AT-biased genotypes of O. sinensis, not the GC-biased H. sinensis, in the stromata, indicating that the sequences of O. sinensis genotypes were present in independent genomes of different fungi [Xiao et al., 2009 [83]; Zhu et al., 2010 [84]; Hu et al., 2013 [31]; Li et al., 2013 [70], 2016a [40], 2020 [14], 2022 [6], 2023c [49]; Jin et al., 2020 [41]; Liu et al., 2020 [42]; Shu et al., 2020 [43]];
- (1-c).
- The GC-biased Genotypes #1 and #2 of O. sinensis cooccur in the stromata of natural C. sinensis. The abundance of the GC-biased genotypes was dynamically altered during C. sinensis maturation [Zhu et al., 2010 [84]];
- (1-d).
- The cooccurrence of GC-biased genomically independent Genotypes #1 and #7 of O. sinensis was detected in the same specimen of natural C. sinensis [Chen et al., 2011 [69]];
- (1-e).
- A species contradiction between the anamorphic inoculants (GC-biased Genotype #1 H. sinensis strains) and the sole teleomorph of AT-biased Genotype #4 of O. sinensis was detected in the fruiting body of cultivated C. sinensis in a product-oriented industrial setting [Wei et al., 2016 [19]];
- (1-f).
- Discovery of Genotypes #13 and #14 of O. sinensis in semi-ejected and fully ejected multicellular heterokaryotic ascospores, respectively, collected from the same C. sinensis samples [Li et al., 2023 [15]];
- (1-g).
- (2)
- Evidence for the differential cooccurrence of heterospecific fungal species in different compartments of the natural C. sinensis insect–fungi complex is as follows:
- (2-a).
- (2-b).
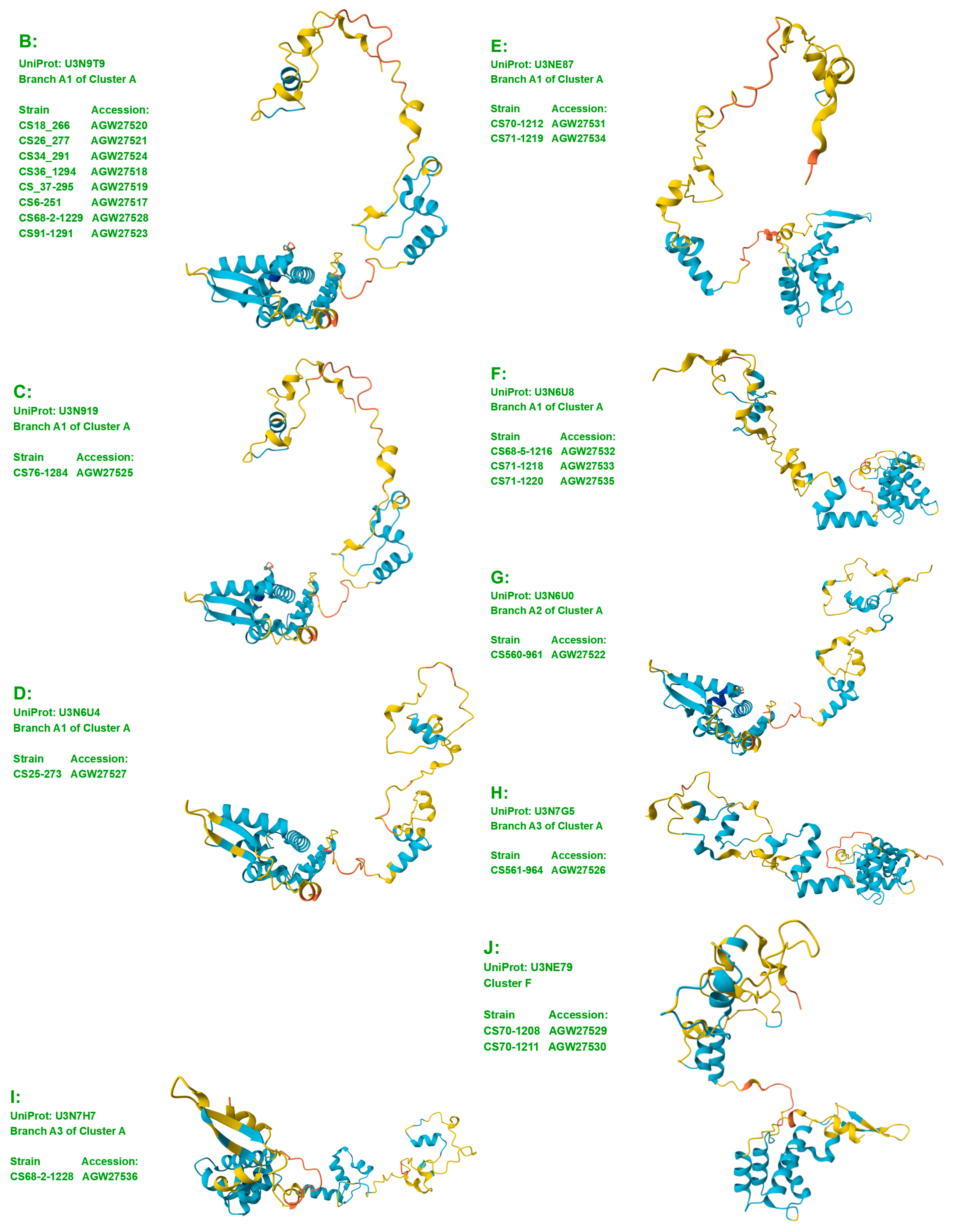
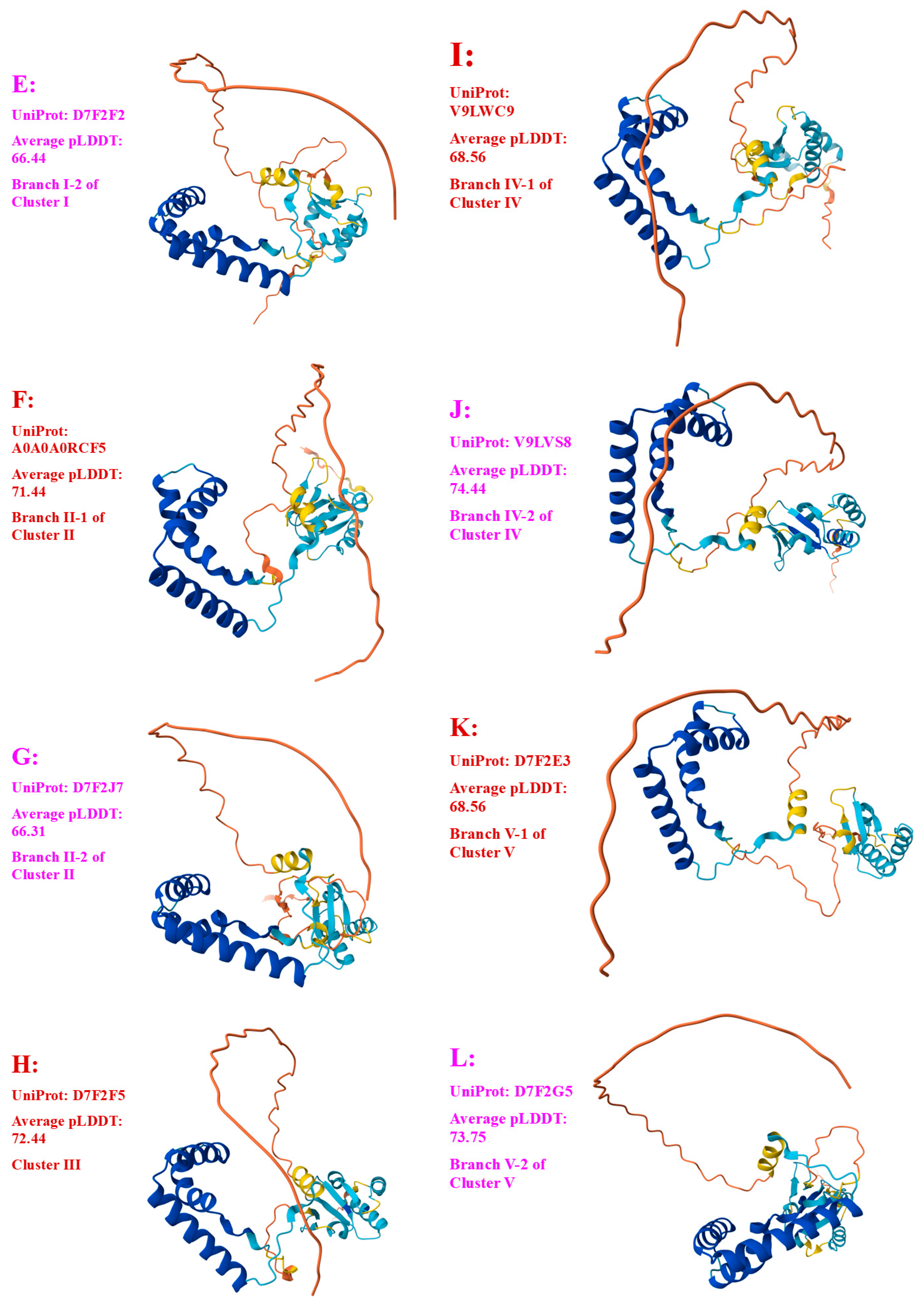
- A good number of C. sinensis isolates contained mutant MAT1-1-1 and MAT1-2-1 proteins, especially those proteins with C- and/or N-terminal truncations that belong to nine and four diverse 3D structural morphs (cf. Figure 4 and Figure 6), respectively. The mutant proteins were either clustered into a separate Bayesian clade or clustered within the main clustering branches in the Bayesian trees (cf. Figure 1 and Figure 2). The MAT1-1-1 and MAT1-2-1 proteins encoded by metatranscriptome assemblies of natural C. sinensis also exhibited either large-segment truncation or sequence variations similar to those observed in wild-type C. sinensis isolates (cf. Figure 7, Figure 8, Figure 9 and Figure 10). Some of the mutant proteins might be produced by heterospecific fungi in impure wild-type C. sinensis isolates and in natural C. sinensis insect–fungi complexes;
- (2-c).
- Discoveries of the formation of the heterospecific Cordyceps-Tolypocladium complex in natural C. sinensis [Engh 1999 [90]; Stensrud et al., 2005 [91], 2007 [92]] and the dual anamorphs of O. sinensis, involving psychrophilic H. sinensis and mesophilic Tolypocladium sinensis [Li 1988 [93]; Chen et al., 2004 [94]; Leung et al., 2006 [95]; Barseghyan et al., 2011 [96]];
- (2-d).
- A close association of psychrophilic H. sinensis and mesophilic S. hepiali (≡P. hepiali) was found in the caterpillar body, stroma, ascospores, and stromal fertile portion, which was densely covered with numerous ascocarps of natural C. sinensis, and in the wild-type C. sinensis complexes, which appeared to be difficult to purify [Dai et al., 1989 [81]; Jiang & Yao 2003 [8]; Chen et al., 2004 [94]; Zhu et al., 2007 [97], 2010 [84]; Yang et al., 2008 [98]; Li et al., 2016 [13], 2023 [15]];
- (2-e).
- Although Genotypes #13–14 are among the 17 genotypes of O. sinensis, these 2 GC-biased genotypes feature precise reciprocal cross substitutions of large DNA segments among two heterospecific parental fungi, namely, H. sinensis and an AB067719-type fungus. The taxonomic position of the AB067719-type fungus is undetermined to date, and more than 900 heterospecific fungal sequences, which are highly homologous to AB067719, have been uploaded to GenBank [Li et al., 2023 [15]]. Chromosomal intertwining and genetic material recombination may occur after plasmogamy and karyogamy of the heterospecific parental fungi under sexual reproduction hybridization or parasexuality, which is characterized by the prevalence of heterokaryosis and results in concerted chromosome loss for transferring–substituting genetic materials without conventional meiosis [Bennett & Johnson 2003 [99]; Sherwood & Bennett 2009 [100]; Bushley et al., 2013 [32]; Seervai et al., 2013 [73]; Nakamura et al., 2019 [74]; Mishra et al., 2021 [78]; Kück et al., 2022 [80]; Li et al., 2023 [15]].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, J.-S.; Halpern, G.M.; Jones, K. The scientific rediscovery of an ancient Chinese herbal medicine: Cordyceps sinensis: Part II. J. Altern. Complem. Med. 1998, 4, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-S.; Li, C.-L.; Tan, N.-Z.; Berger, J.L.; Prolla, T.A. Combined use of whole-gene expression profiling technology and mouse lifespan test in anti-aging herbal product study. In Proceedings of the 2011 New TCM Products Innovation and Industrial Development Summit, Hangzhou, China, 27 November 2011; pp. 443–448. Available online: https://xueshu.baidu.com/usercenter/paper/show?paperid=08341c17fa58c8f85584b92572b90f75&site=xueshu_se (accessed on 30 January 2025).
- Ren, Y.; Wan, D.-G.; Lu, X.-M.; Guo, J.-L. The study of scientific name discussion for TCM Cordyceps. LisShenzhen Med. Mater. Medica Res. 2013, 24, 2211–2212. [Google Scholar]
- Zhang, Y.-J.; Zhang, S.; Li, Y.-L.; Ma, S.-L.; Wang, C.-S.; Xiang, M.-C.; Liu, X.; An, Z.-Q.; Xu, J.-P.; Liu, X.-Z. Phylogeography and evolution of a fungal–insect association on the Tibetan Plateau. Mol. Ecol. 2014, 23, 5337–5355. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-L.; St Leger, R.J. Chapter Seven—Insect Immunity to Entomopathogenic Fungi; Lovett, B., St Leger, R.J., Eds.; Advanc Genet; Academic Press: Cambridge, MA, USA, 2016; Volume 94, pp. 251–285. [Google Scholar]
- Li, Y.-L.; Li, X.-Z.; Yao, Y.-S.; Xie, W.-D.; Zhu, J.-S. Molecular identification of Ophiocordyceps sinensis genotypes and the indiscriminate use of the Latin name for the multiple genotypes and the natural insect-fungi complex. Am. J. BioMed Sci. 2022, 14, 115–135. [Google Scholar] [CrossRef]
- Li, M.-M.; Zhang, J.-H.; Qin, Q.-L.; Zhang, H.; Li, X.; Wang, H.-T.; Meng, Q. Transcriptome and Metabolome Analyses of Thitarodes xiaojinensis in Response to Ophiocordyceps sinensis Infection. Microorganisms 2023, 11, 2361. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yao, Y.-J. A review for the debating studies on the anamorph of Cordyceps sinensis. Mycosistema 2003, 22, 161–176. [Google Scholar]
- Zhang, Y.-J.; Sun, B.-D.; Zhang, S.; Wang, M.; Liu, X.-Z.; Gong, W.-F. Mycobiotal investigation of natural Ophiocordyceps sinensis based on culture-dependent investigation. Mycosistema 2010, 29, 518–527. [Google Scholar]
- Zhang, S.-W.; Cen, K.; Liu, Y.; Zhou, X.-W.; Wang, C.-S. Metatranscriptomics analysis of the fruiting caterpillar fungus collected from the Qinghai-Tibetan plateau. Sci. Sinica Vitae 2018, 48, 562–570. [Google Scholar]
- Xia, F.; Liu, Y.; Shen, G.-L.; Guo, L.-X.; Zhou, X.-W. Investigation and analysis of microbiological communities in natural Ophiocordyceps sinensis. Can. J. Microbiol. 2015, 61, 104–111. [Google Scholar] [CrossRef]
- Guo, M.-Y.; Liu, Y.; Gao, Y.-H.; Jin, T.; Zhang, H.-B.; Zhou, X.-W. Identification and bioactive potential of endogenetic fungi isolated from medicinal caterpillar fungus Ophiocordyceps sinensis from Tibetan Plateau. Int. J. Agric. Biol. 2017, 19, 307–313. [Google Scholar] [CrossRef]
- Li, Y.-L.; Yao, Y.-S.; Zhang, Z.-H.; Xu, H.-F.; Liu, X.; Ma, S.-L.; Wu, Z.-M.; Zhu, J.-S. Synergy of fungal complexes isolated from the intestines of Hepialus lagii larvae in increasing infection potency. J. Fungal Res. 2016, 14, 96–112. [Google Scholar]
- Li, X.-Z.; Li, Y.-L.; Yao, Y.-S.; Xie, W.-D.; Zhu, J.-S. Further discussion with Li et al. (2013, 2019) regarding the “ITS pseudogene hypothesis” for Ophiocordyceps sinensis. Mol. Phylogenet. Evol. 2020, 146, 106728. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Li, X.-Z.; Yao, Y.-S.; Wu, Z.-M.; Gao, L.; Tan, N.-Z.; Lou, Z.-Q.; Xie, W.-D.; Wu, J.-Y.; Zhu, J.-S. Differential cooccurrence of multiple genotypes of Ophiocordyceps sinensis in the stromata, stromal fertile portion (ascocarps) and ascospores of natural Cordyceps sinensis. PLoS ONE 2023, 18, e0270776. [Google Scholar] [CrossRef]
- Zhong, X.; Gu, L.; Wang, H.-Z.; Lian, D.-H.; Zheng, Y.-M.; Zhou, S.; Zhou, W.; Gu, J.; Zhang, G.; Liu, X. Profile of Ophiocordyceps sinensis transcriptome and differentially expressed genes in three different mycelia, sclerotium and fruiting body developmental stages. Fungal Biol. 2018, 122, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Zhang, J.; Chen, F.; Dong, C.; Qin, Q.; Li, X.; Wang, H.; Zhang, H.; Meng, Q. Unveiling mycoviral diversity in Ophiocordyceps sinensis through transcriptome analyses. Front. Microbiol. 2024, 15, 1493365. [Google Scholar] [CrossRef]
- Wei, X.-L.; Yin, X.-C.; Guo, Y.-L.; Shen, N.-Y.; Wei, J.-C. Analyses of molecular systematics on Cordyceps sinensis and its related taxa. Mycosystema 2006, 25, 192–202. [Google Scholar]
- Wei, J.-C.; Wei, X.-L.; Zheng, W.-F.; Guo, W.; Liu, R.-D. Species identification and component detection of Ophiocordyceps sinensis cultivated by modern industry. Mycosystema 2016, 35, 404–410. [Google Scholar]
- Sung, G.-H.; Hywel-Jones, N.L.; Sung, J.-M.; Luangsa-ard, J.J.; Shrestha, B.; Spatafora, J.W. Phylogenetic classification of Cordyceps and the clavicipitaceous fungi. Stud. Mycol. 2007, 57, 5–59. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Li, E.-W.; Wang, C.-S.; Li, Y.-L.; Liu, X.-Z. Ophiocordyceps sinensis, the flagship fungus of China: Terminology, life strategy and ecology. Mycology 2012, 3, 2–10. [Google Scholar] [CrossRef]
- Yao, Y.-S.; Zhu, J.-S. Indiscriminate use of the Latin name for natural Cordyceps sinensis and Ophiocordyceps sinensis fungi. Chin. J. Chin. Mater. Med. 2016, 41, 1316–1366. [Google Scholar]
- Zhang, S.; Zhang, Y.-J.; Shrestha, B.; Xu, J.-P.; Wang, C.-S.; Liu, X.-Z. Ophiocordyceps sinensis and Cordyceps militaris: Research advances, issues and perspectives. Mycosystema 2013, 32, 577–597. [Google Scholar]
- Hawksworth, D.L.; Crous, P.W.; Redhead, S.A.; Reynolds, D.R.; Samson, R.A.; Seifert, K.A.; Taylor, J.W.; Wingfield, M.J.; Abaci, Ö.; Aime, C.; et al. The Amsterdam declaration on fungal nomenclature. IMA Fungus 2011, 2, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Debuchy, R.; Turgeo, B.G. Mating-Type Structure, Evolution, and Function in Euascomycetes. In Growth, Differentiation and Sexuality; Kües, U., Fischer, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 293–323. [Google Scholar]
- Jones, S.K.; Bennett, R.J. Fungal mating pheromones: Choreographing the dating game. Fungal Genet. Biol. 2011, 48, 668–676. [Google Scholar] [CrossRef]
- Zheng, P.; Wang, C.-S. Sexuality Control and Sex Evolution in Fungi. Sci. Sin. Vitae 2013, 43, 1090–1097. [Google Scholar]
- Wilson, A.M.; Wilken, P.M.; van der Nest, M.A.; Steenkamp, E.T.; Wingfield, M.J.; Wingfield, B.D. Homothallism: An umbrella term for describing diverse sexual behaviours. IMA Fungus 2015, 6, 207–214. [Google Scholar] [CrossRef]
- Holliday, J.; Cleaver, M. Medicinal value of the caterpillar fungi species of the genus Cordyceps (Fr.) Link (Ascomycetes). A review. Int. J. Med. Mushrooms 2008, 10, 219–234. [Google Scholar] [CrossRef]
- Stone, R. Improbable partners aim to bring biotechnology to a Himalayan kingdom. Science 2010, 327, 940–941. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Y.-J.; Xiao, G.-H.; Zheng, P.; Xia, Y.-L.; Zhang, X.-Y.; St Leger, R.J.; Liu, X.-Z.; Wang, C.-S. Genome survey uncovers the secrets of sex and lifestyle in caterpillar fungus. Chin. Sci. Bull. 2013, 58, 2846–2854. [Google Scholar] [CrossRef]
- Bushley, K.E.; Li, Y.; Wang, W.-J.; Wang, X.-L.; Jiao, L.; Spatafora, J.W.; Yao, Y.-J. Isolation of the MAT1-1 mating type idiomorph and evidence for selfing in the Chinese medicinal fungus Ophiocordyceps sinensis. Fungal Biol. 2013, 117, 599–610. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xu, L.-L.; Zhang, S.; Liu, X.-Z.; An, Z.-Q.; Wang, M.; Guo, Y.-L. Genetic diversity of Ophiocordyceps sinensis, a medicinal fungus endemic to the Tibetan Plateau: Implications for its evolution and conservation. BMC Evol. Biol. 2009, 9, 290. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.-J.; Liu, X.-Z.; Wen, H.-A.; Wang, M.; Liu, D.-S. Cloning and analysis of the MAT1-2-1 gene from the traditional Chinese medicinal fungus Ophiocordyceps sinensis. Fungal Biol. 2011, 115, 708–714. [Google Scholar]
- Zhang, S.; Zhang, Y.-J. Molecular evolution of three protein-coding genes in the Chinese caterpillar fungus Ophiocordyceps sinensis. Microbiol. China 2015, 42, 1549–1560. [Google Scholar]
- Li, X.-Z.; Li, Y.-L.; Zhu, J.-S. Differential transcription of mating-type genes during sexual reproduction of natural Cordyceps sinensis. Chin. J. Chin. Mater. Medica 2023, 48, 2829–2840. [Google Scholar] [CrossRef]
- Li, X.-Z.; Xiao, M.-J.; Li, Y.-L.; Gao, L.; Zhu, J.-S. Mutations and differential transcription of mating-type and pheromone receptor genes in Hirsutella sinensis and the natural Cordyceps sinensis insect—fungi complex. Biology 2024, 13, 632. [Google Scholar] [CrossRef] [PubMed]
- China Ministry of Agriculture and Rural Affairs. Announcement (No. 15 of 2021) of National Forestry and Grassland Administration: List of National Key Protected Wild Plants. 7 September 2021. Available online: https://www.forestry.gov.cn/c/www/gkml/11057.jhtml (accessed on 30 January 2025).
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Li, Y.; Hsiang, T.; Yang, R.-H.; Hu, X.-D.; Wang, K.; Wang, W.-J.; Wang, X.-L.; Jiao, L.; Yao, Y.-J. Comparison of different sequencing and assembly strategies for a repeat-rich fungal genome, Ophiocordyceps sinensis. J. Microbiol. Methods 2016, 128, 1–6. [Google Scholar] [CrossRef]
- Jin, L.-Q.; Xu, Z.-W.; Zhang, B.; Yi, M.; Weng, C.-Y.; Lin, S.; Wu, H.; Qin, X.-T.; Xu, F.; Teng, Y.; et al. Genome sequencing and analysis of fungus Hirsutella sinensis isolated from Ophiocordyceps sinensis. AMB Expr. 2020, 10, 105. [Google Scholar] [CrossRef]
- Liu, J.; Guo, L.-N.; Li, Z.-W.; Zhou, Z.; Li, Z.; Li, Q.; Bo, X.-C.; Wang, S.-Q.; Wang, J.-L.; Ma, S.-C.; et al. Genomic analyses reveal evolutionary and geologic context for the plateau fungus Ophiocordyceps sinensis. Clin. Med. 2020, 15, 107–119. [Google Scholar] [CrossRef]
- Shu, R.-H.; Zhang, J.-H.; Meng, Q.; Zhang, H.; Zhou, G.-L.; Li, M.-M.; Wu, P.-P.; Zhao, Y.-N.; Chen, C.; Qin, Q.-L. A new high-quality draft genome assembly of the Chinese cordyceps Ophiocordyceps sinensis. Genome Biol. Evol. 2020, 12, 1074–1079. [Google Scholar] [CrossRef]
- Liu, Z.-Q.; Lin, S.; Baker, P.J.; Wu, L.-F.; Wang, X.-R.; Wu, H.; Xu, F.; Wang, H.-Y.; Brathwaite, M.E.; Zheng, Y.-G. Transcriptome sequencing and analysis of the entomopathogenic fungus Hirsutella sinensis isolated from Ophiocordyceps sinensis. BMC Genom. 2015, 16, 106–123. [Google Scholar] [CrossRef]
- Xiang, L.; Li, Y.; Zhu, Y.; Luo, H.; Li, C.; Xu, X.; Sun, C.; Song, J.-Y.; Shi, L.-H.; He, L.; et al. Transcriptome analysis of the Ophiocordyceps sinensis fruiting body reveals putative genes involved in fruiting body development and cordycepin biosynthesis. Genomics 2014, 103, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.-H.; Yang, D.-R.; Jiang, J.-J.; Zhang, Q.-J.; Liu, Y.; Liu, Y.-L.; Zhang, Y.; Zhang, H.-B.; Shi, C.; Tong, Y.; et al. The caterpillar fungus, Ophiocordyceps sinensis, genome provides insights into highland adaptation of fungal pathogenicity. Sci. Rep. 2017, 7, 1806. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Li, Y.-L.; Gao, L.; Yao, Y.-S.; Wu, Z.-M.; Lou, Z.-Q.; Xie, W.-D.; Wu, J.-Y.; Zhu, J.-S. Altered GC- and AT-biased genotypes of Ophiocordyceps sinensis in the stromal fertile portions and ascospores of natural Cordyceps sinensis. PLoS ONE 2023, 18, e0286865. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- David, A.; Islam, S.; Tankhilevich, E.; Sternberg, M.J.E. The AlphaFold Database of Protein Structures: A Biologist’s Guide. J. Mol. Biol. 2022, 434, 167336. [Google Scholar] [CrossRef] [PubMed]
- Rettie, S.A.; Campbell, K.V.; Bera, A.K.; Kang, A.; Kozlov, S.; De La Cruz, J.; Adebomi, V.; Zhou, G.; DiMaio, F.; Ovchinnikov, S.; et al. Cyclic peptide structure prediction and design using AlphaFold. bioRxiv 2023. Preprint. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Monzon, V.; Haft, D.H.; Bateman, A. Folding the unfoldable: Using AlphaFold to explore spurious proteins. Bioinform. Adv. 2022, 1, vbab043. [Google Scholar] [CrossRef]
- Xu, T.; Xu, Q.; Li, J.-Y. Toward the appropriate interpretation of Alphafold2. Front. Artif. Intell. 2023, 6, 1149748. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, K.; Kmiecik, S. Integrating AlphaFold pLDDT Scores into CABS-flex for enhanced protein flexibility simulations. Comput. Struct. Biotechnol. J. 2024, 30, 4350–4356. [Google Scholar] [CrossRef]
- Deleage, G.; Roux, B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. Des. Sel. 1987, 1, 289–294. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; Chapter 52; pp. 571–607. [Google Scholar]
- Peters, C.; Elofsson, A. Why is the biological hydrophobicity scale more accurate than earlier experimental hydrophobicity scales? Proteins 2014, 82, 2190–2198. [Google Scholar] [CrossRef]
- Simm, S.; Einloft, J.; Mirus, O.; Schleiff, E. 50 years of amino acid hydrophobicity scales: Revisiting the capacity for peptide classification. Biol. Res. 2016, 49, 31. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Z.; Li, Y.-L.; Wang, Y.-N.; Zhu, J.-S. Translations of mutant repetitive genomic sequences in Hirsutella sinensis and changes in secondary structures and functional specifications of the encoded proteins. Int. J. Mol. Sci. 2024, 25, 11178. [Google Scholar] [CrossRef]
- Jacobsen, S.; Wittig, M.; Pöggeler, S. Interaction Between Mating-Type Proteins From the Homothallic Fungus Sordaria macrospora. Curr. Genet. 2002, 41, 150–158. [Google Scholar] [CrossRef]
- Rams, B.; Kück, U. The Penicillium chrysogenum tom1 gene a major target of transcription factor MAT1-1-1 encodes a nuclear protein involved in sporulation. Front. Fungal Biol. 2022, 3, 937023. [Google Scholar] [CrossRef]
- Zhu, J.-S.; Gray, G.M. Renaturative catalytic blotting of enzyme proteins. In Protein Blotting: A practical Approach (IRL Series); Dunbar, B.S., Ed.; Oxford University Press: Oxford, UK, 1994. [Google Scholar] [CrossRef]
- Turgeon, B.G.; Yoder, O.C. Proposed nomenclature for mating type genes of filamentous ascomycetes. Fungal Genet. Biol. 2000, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, N.; Zang, M. Morphological and phylogenetic studies on Cordyceps sinensis distributed in southwestern China. Mycoscience 2001, 42, 567–574. [Google Scholar] [CrossRef]
- Chen, C.-S.; Hseu, R.-S.; Huang, C.-T. Quality control of Cordyceps sinensis teleomorph, anamorph, and Its products. In Quality Control of Herbal Medicines and Related Areas; Shoyama, Y., Ed.; InTech: Rijeka, Croatia, 2011; Chapter 12; pp. 223–238. Available online: www.intechopen.com (accessed on 30 January 2025).
- Li, Y.; Jiao, L.; Yao, Y.-J. Non-concerted ITS evolution in fungi, as revealed from the important medicinal fungus Ophiocordyceps sinensis. Mol. Phylogenet. Evol. 2013, 68, 373–379. [Google Scholar] [CrossRef]
- Mao, X.-M.; Zhao, S.-M.; Cao, L.; Yan, X.; Han, R.-C. The morphology observation of Ophiocordyceps sinensis from different origins. J. Environ. Entomol. 2013, 35, 343–353. [Google Scholar]
- Pfennig, K.S. Facultative Mate Choice Drives Adaptive Hybridization. Science 2007, 318, 965–967. [Google Scholar] [CrossRef]
- Seervai, R.N.H.; Jones, S.K.; Hirakawa, M.P.; Porman, A.M.; Bennett, R.J. Parasexuality and ploidy change in Candida tropicalis. Eukaryot. Cell 2013, 12, 1629–1640. [Google Scholar] [CrossRef]
- Nakamura, N.; Tanaka, C.; Takeuchi-Kaneko, Y. Transmission of antibiotic-resistance markers by hyphal fusion suggests partial presence of parasexuality in the root endophytic fungus Glutinomyces brunneus. Mycol. Prog. 2019, 18, 453–462. [Google Scholar] [CrossRef]
- Du, X.-H.; Wu, D.-M.; Kang, H.; Wang, H.-C.; Xu, N.; Li, T.-T.; Chen, K.-L. Heterothallism and potential hybridization events inferred for twenty-two yellow morel species. IMA Fungus 2020, 11, 4. [Google Scholar] [CrossRef]
- Hėnault, M.; Marsit, S.; Charron, G.; Landry, C.R. The effect of hybridization on transposable element accumulation in an undomesticated fungal species. eLife 2020, 9, e60474. [Google Scholar] [CrossRef]
- Samarasinghe, H.; You, M.; Jenkinson, T.S.; Xu, J.-P.; James, T.Y. Hybridization Facilitates Adaptive Evolution in Two Major Fungal Pathogens. Genes 2020, 11, 101. [Google Scholar] [CrossRef]
- Mishra, A.; Forche, A.; Anderson, M.Z. Parasexuality of Candida Species. Front. Cell. Infect. Microbiol. 2021, 11, 796929. [Google Scholar] [CrossRef]
- Steensels, J.; Gallone, B.; Verstrepen, K.J. Interspecific hybridization as a driver of fungal evolution and Adaptation. Nat. Rev. Microbiol. 2021, 19, 485–500. [Google Scholar] [PubMed]
- Kück, U.; Bennett, R.J.; Wang, L.; Dyer, P.S. Editorial: Sexual and Parasexual Reproduction of Human Fungal Pathogens. Front. Cell. Infect. Microbiol. 2022, 12, 934267. [Google Scholar] [CrossRef]
- Dai, R.-Q.; Lan, J.-L.; Chen, W.-H.; Li, X.-M.; Chen, Q.-T.; Shen, C.-Y. Discovery of a new fungus Paecilomyces hepiali Chen & Dai. Acta Agricult. Univ. Pekin. 1989, 15, 221–224. [Google Scholar]
- Wang, Y.-B.; Wang, Y.; Fan, Q.; Duan, D.-E.; Zhang, G.-D.; Dai, R.-Q.; Dai, Y.-D.; Zeng, W.-B.; Chen, Z.-H.; Li, D.-D.; et al. Multigene phylogeny of the family Cordycipitaceae (Hypocreales): New taxa and the new systematic position of the Chinese cordycipitoid fungus Paecilomyces hepiali. Fungal Divers 2020, 103, 1. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, J.-P.; Zhu, P.; Cheng, K.-D.; He, H.-X.; Zhu, H.-X.; Wang, Q. Non-support of species complex hypothesis of Cordyceps sinensis by targeted rDNA-ITS sequence analysis. Mycosystema 2009, 28, 724–730. [Google Scholar]
- Zhu, J.-S.; Gao, L.; Li, X.-H.; Yao, Y.-S.; Zhou, Y.-J.; Zhao, J.-Q.; Zhou, Y.-J. Maturational alterations of oppositely orientated rDNA and differential proliferations of CG:AT-biased genotypes of Cordyceps sinensis fungi and Paecilomyces hepiali in natural C. sinensis. Am. J. Biomed. Sci. 2010, 2, 217–238. [Google Scholar] [CrossRef]
- Meng, Q.; Yu, H.-Y.; Zhang, H.; Zhu, W.; Wang, M.-L.; Zhang, J.-H.; Zhou, G.-L.; Li, X.; Qin, Q.-L.; Hu, S.-N.; et al. Transcriptomic insight into the immune defenses in the ghost moth, Hepialus xiaojinensis, during an Ophiocordyceps sinensis fungal infection. Insect Biochem. Mol. Biol. 2015, 64, 1–15. [Google Scholar] [CrossRef]
- Wang, Y.; Stata, M.; Wang, W.; Stajich, J.E.; White, M.M.; Moncalvo, J.M. Comparative genomics reveals the core gene toolbox for the fungus-insect symbiosis. mBio 2018, 9, e00636-18. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Liu, Q.; Li, Q.-P.; Qian, Z.-M.; Zhang, X.-L.; Li, K.; Li, W.-J.; Dong, C.-H. Developmental transcriptomics of Chinese cordyceps reveals gene regulatory network and expression profiles of sexual development-related genes. BMC Genom. 2019, 20, 337. [Google Scholar] [CrossRef]
- Zhao, Y.-N.; Zhang, J.-H.; Meng, Q.; Zhang, H.; Zhou, G.-L.; Li, M.-M.; Wu, P.-P.; Shu, R.-H.; Gao, X.-X.; Guo, L.; et al. Transcriptomic analysis of the orchestrated molecular mechanisms underlying fruiting body initiation in Chinese cordyceps. Gene 2020, 763, 145061. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Tong, X.-X.; He, C.-Y.; Bai, J.; Wang, F.; Guo, J.-L. Comparison of endogenetic microbial community diversity between wild Cordyceps sinensis, artificial C. sinensis and habitat soil. Chin. J. Chin. Mater. Medica 2021, 46, 3106–3115. [Google Scholar]
- Engh, I.B. Molecular Phylogeny of the Cordyceps-Tolypocladium Complex. Ph.D. Thesis, Department of Biology, University of Oslo, Oslo, Norway, 1999. [Google Scholar]
- Stensrud, Ø.; Hywel-Jones, N.L.; Schumacher, T. Towards a phylogenetic classification of Cordyceps: ITS nrDNA sequence data confirm divergent lineages and paraphyly. Mycol. Res. 2005, 109, 41–56. [Google Scholar] [CrossRef]
- Stensrud, Ø.; Schumacher, T.; Shalchian-Tabrizi, K.; Svegardenib, I.B.; Kauserud, H. Accelerated nrDNA evolution and profound AT bias in the medicinal fungus Cordyceps sinensis. Mycol. Res. 2007, 111, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-L. A study of Tolypocladium sinense C.L. Li. sp. nov. and cyclosporin production. Acta Mycol. Sinica 1988, 7, 93–98. [Google Scholar]
- Chen, Y.-Q.; Hu, B.; Xu, F.; Zhang, W.; Zhou, H.; Qu, L.-H. Genetic variation of Cordyceps sinensis, a fruit-body-producing entomopathogenic species from different geographical regions in China. FEMS Microbiol Lett. 2004, 230, 153–158. [Google Scholar] [CrossRef]
- Leung, P.-H.; Zhang, Q.-X.; Wu, J.-Y. Mycelium cultivation, chemical composition and antitumour activity of a Tolypocladium sp. fungus isolated from wild Cordyceps sinensis. J. Appl. Microbiol. 2006, 101, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Barseghyan, G.S.; Holliday, J.C.; Price, T.C.; Madison, L.M.; Wasser, S.P. Growth and cultural-morphological characteristics of vegetative mycelia of medicinal caterpillar fungus Ophiocordyceps sinensis G.H. Sung et al. (Ascomycetes) Isolates from Tibetan Plateau (P. R. China). Int. J. Med. Mushrooms 2011, 13, 565–581. [Google Scholar] [CrossRef]
- Zhu, J.-S.; Guo, Y.-L.; Yao, Y.-S.; Zhou, Y.-J.; Lu, J.-H.; Qi, Y.; Chen, W.; Zheng, T.-Y.; Zhang, L.; Wu, Z.-M.; et al. Maturation of Cordyceps sinensis associates with co-existence of Hirsutella sinensis and Paecilomyces hepiali DNA and dynamic changes in fungal competitive proliferation predominance and chemical profiles. J. Fungal Res. 2007, 5, 214–224. [Google Scholar]
- Yang, J.-L.; Xiao, W.; He, H.-X.; Zhu, H.-X.; Wang, S.-F.; Cheng, K.-D.; Zhu, P. Molecular phylogenetic analysis of Paecilomyces hepiali and Cordyceps sinensis. Acta Pharmaceut. Sinica 2008, 43, 421–426. [Google Scholar] [CrossRef]
- Bennett, R.J.; Johnson, A.D. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J. 2003, 22, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, R.K.; Bennett, R.J. Fungal meiosis and parasexual reproduction--lessons from pathogenic yeast. Curr. Opin. Microbiol. 2009, 12, 599–607. [Google Scholar] [CrossRef] [PubMed]

 very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
 very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).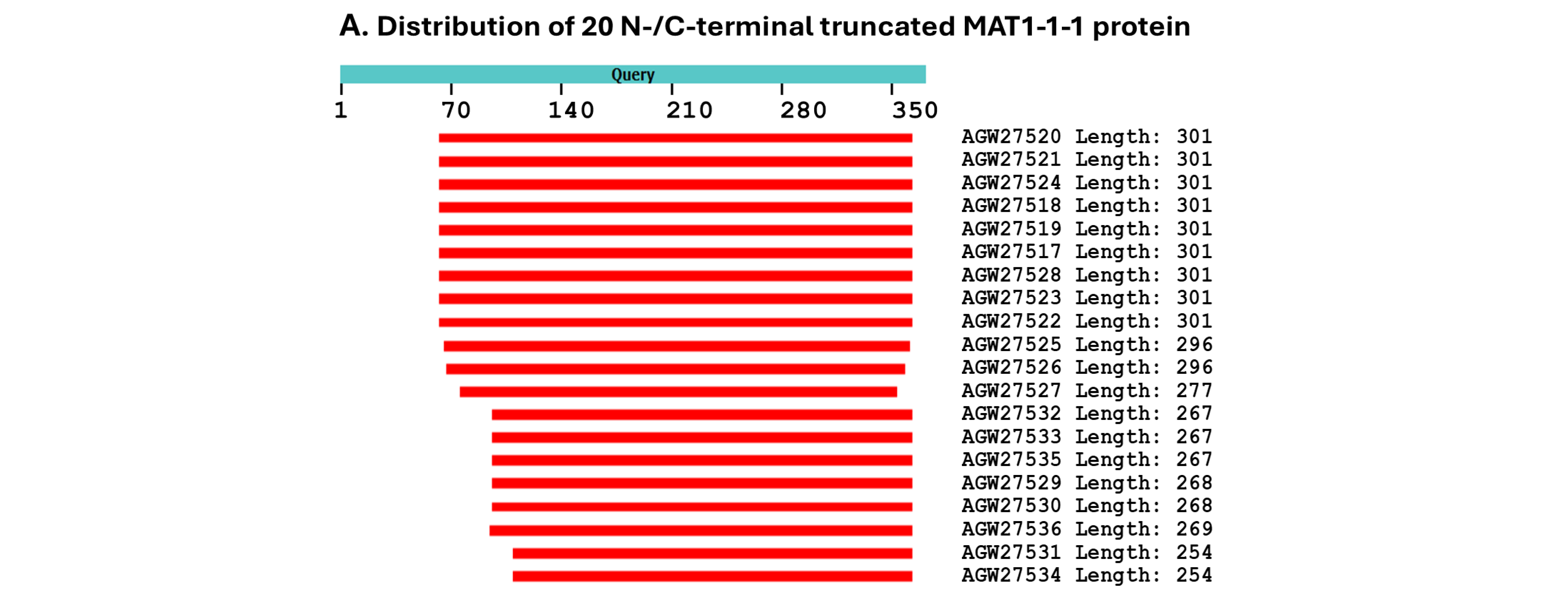
 very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
 very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).
very high (pLDDT > 90), high (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AlphaFold UniProt Code (Bayesian Cluster/Branch *) | Strain/Isolate Number (GenBank Accession Number) |
|---|---|
| U3N942 (A1) | CS68-2-1229 (AGW27560) (AGW27528), GS09_111 (ALH24945), GS09_131 (ALH24947), ID10_1 (ALH24954), IOZ07 (KAF4512729), NP10_1 (ALH24955), NP10_2 (ALH24956), QH07_188 (ALH24957), QH07_197 (ALH24958), QH09_37 (ALH24968), QH09_46 (ALH24969), QH09_56 (ALH24970), QH09_66 (ALH24971), QH09_78 (ALH24972), QH09_93 (ALH24973), QH09_122 (ALH24959), QH09_131 (ALH24960), QH09_151 (ALH24961), QH09_20L (ALH24965), QH09_33L (ALH24967), QH10_1 (ALH24974), QH10_4 (ALH24975), QH10_7 (ALH24976), SC09_21 (ALH24987), SC09_36 (ALH24988), SC09_37 (ALH24989), SC09_47 (ALH24990), SC09_57 (ALH24991), SC09_77 (ALH24993), SC09_107 (ALH24978), SC09_117 (ALH24979), SC09_128 (ALH24980), SC09_147 (ALH24981), SC09_157 (ALH24982), SC09_167 (ALH24983), SC09_180 (ALH24984), SC09_190 (ALH24985), SC09_200 (ALH24986), SC10_18 (ALH24996), SC10_21 (ALH24997), SC10_4 (ALH24998), XZ05_3 (ALH25002), XZ05_7 (ALH25004), XZ05_12 (ALH25000), XZ06_124 (ALH25006), XZ06_152 (ALH25007), XZ07_108 (ALH25009), XZ07_133 (ALH25010), XZ07_154 (ALH25011), XZ07_166 (ALH25012), XZ07_176 (ALH25013), XZ07_180 (ALH25014), XZ08_4 (ALH25018), XZ08_10 (ALH25015), XZ08_24 (ALH25016), XZ08_26 (ALH25017), XZ08_56 (ALH25019), XZ08_59 (ALH25020), XZ08_A1 (ALH25021), XZ08_B1 (ALH25022), XZ09_4 (ALH25029), XZ09_46 (ALH25030), XZ09_48 (ALH25031), XZ09_59 (ALH25032), XZ09_71 (ALH25033), XZ09_80 (ALH25055), XZ09_106 (ALH25024), XZ09_113 (ALH25025), XZ09_118 (ALH25026), XZ09_15 (ALH25027), XZ09_32 (ALH25028), XZ10_7 (ALH25038), XZ10_15 (ALH25035), XZ10_17 (ALH25036), XZ10_23 (ALH25037), XZ12_1 (ALH25056), XZ12_33 (ALH25058), XZ12_43 (ALH25059), YN07_6 (ALH25039), YN07_8 (ALH25040), YN09_3 (ALH25044), YN09_72 (ALH25049), YN09_81 (ALH25050), YN09_85 (ALH25051), YN09_89 (ALH25052), YN09_96 (ALH25053),YN09_101 (ALH25041), YN09_140 (ALH25042) |
| A0A0N9QMM1 (A1) | GS09_121 (ALH24946), GS09_201 (ALH24949), GS09_225 (ALH24950), SC09_1 (ALH24977) |
| T5A511 (A1) | Co18 (EQK97643) (KE657544 410←1519 and ANOV01017390 410←1519) |
| A0A0N9R5B3 (A2) | SC09_65 (ALH24992) |
| A0A0N7G849 (A2) | SC09_97 (ALH24995) |
| A0A0N9QUF3 (A3) | GS09_143 (ALH24948) |
| A0A0N9R4V2 (A3) | YN09_61 (ALH25047) |
| A0A0N9QMS9 (B) | YN09_6 (ALH25046), YN09_22 (ALH25043), YN09_51 (ALH25045), YN09_64 (ALH25048) |
| A0A0N7G845 (C) | GS09_229 (ALH24951), GS09_281 (ALH24952), GS09_311 (ALH25054), GS10_1 (ALH24953), QH09_164 (ALH24962), QH09_173 (ALH24963), QH09_201 (ALH24964), QH09_210 (ALH24966), SC09_87 (ALH24994) |
| A0A0N9QUK2 (D1) | XZ05_8 (ALH25005) |
| A0A0N9QMT4 (D2) | XZ07_H2 (ALH24999), XZ12_16 (ALH25057) |
| A0A0N9QMR3 (E1) | XZ06_260 (ALH25008), XZ09_100 (ALH25023) |
| A0A0N9QMS4 (E2) | XZ09_95 (ALH25034) |
| A0A0N7G850 (E3) | XZ05_6 (ALH25003) |
| A0A0N9R4Q4 (E4) | XZ05_2 (ALH25001) |
| AlphaFold UniProt Code (Bayesian Cluster/Branch **) | Strain/Isolate Number (GenBank Accession Number) |
|---|---|
| D7F2E9 (I-1) | CS2 (AEH27625) (ACV60400), CS26-277 (AGW27541), CS36-1294 (AGW27538), CS37-295 (AGW27539), SC-2 (ACV60395), SC-4 (ACV60396), SC-5 (ACV60398), SC-7 (ACV60397), SC09-37 (AFH35019), SC09_47 (AFX66423), SC09_57 (AFX66424), SC09_77 (AFX66426), SC09_97 (AFX66428), XZ05_7 (AFX66442), XZ05_12 (AFX66444), XZ06_152 (AFX66445),XZ07_11 (AFX66447), XZ07_46 (AFX66448), XZ09_106 (AFX66464), XZ09_15 (AFX66455), YN09_101 (AFX66482), YN09_72 (AFX66477), XZ09_113 (AFX66465), XZ-LZ06-1 (ACV60369), XZ-LZ06-7 (ACV60370), XZ-LZ06-21 (ACV60371), XZ-LZ06-108 (ACV60373), XZ-LZ07-30 (ACV60377), XZ-LZ07-108 (ACV60379), XZ-ML-191 (ACV60376), YN-1 (ACV60390), YN-5 (ACV60392), YN-6 (ACV60393), YN-8 (ACV60394), YN09_81 (AFX66478), YN09_85 (AFX66479), YN09_89 (AFX66480) |
| T5AF56 (I-1) | Co18 (EQL04085) (ANOV01000063 9329→10182) |
| V9LW10 (I-2) | SC09_200 (AFX66437) |
| D7F2H1 (I-2) | YN-4 (ACV60391) |
| D7F2F2 (I-2) | XZ-LZ06-61 (ACV60372) |
| A0A0A0RCF5 (II-1) | XZ12_16 (AIV43040) |
| D7F2J7 (II-2) | XZ05_8 (AFX66443), XZ06-124 (AFH35020), XZ-LZ07-H2 (ACV60418), XZ-LZ07-H1 (ACV60417) |
| D7F2F5 (III) | XZ05_2 (AFX66441), XZ06_260 (AFX66446), XZ09_80 (AFX66461), XZ09_95 (AFX66462), XZ09_100 (AFX66463), XZ-LZ05-6 (ACV60415), XZ-SN-44 (ACV60375), |
| V9LWC9 (IV-1) | YN09_64 (AFX66476) |
| V9LVS8 (IV-2) | YN09_6 (AFX66472), YN09_22 (AFX66473), YN09_51 (AFX66474) |
| D7F2E3 (V-1) | GS09_111 (AFX66388), CS560-961 (AGW275424), QH09-93 (AFH35018), XZ-NQ-154 (ACV60363), XZ-NQ-155 (ACV6036) |
| D7F2G5 (V-2) | QH-YS-199 (ACV60385) |
| D7F2H9 (V-2) | SC-3 (ACV60399) |
| V9LW71 (V-2) | QH09_11 (AFX66401) |
| V9LVU8 (V-2) | YN09_61 (AFX66475) |
| V9LWG5 (V-2) | ID10_1 (AFX66484) |
| U3N6V5 (V-2) | CS6-251 (AGW27537) |
| ‡ |
NP10_1 (AFX66485), NP10_2 (AFX66486), YN09_3 (AFX66471), YN09_96 (AFX66481), YN09_140 (AFX66483) |
| Accession Number | % Similarity to AGW27560 | Amino Acid Residue Substitution | Bayesian Cluster | 3D Structure Model | AlphaFold UniProt Code | |||
|---|---|---|---|---|---|---|---|---|
| Conservative | Nonconservative | Branch | Cluster | |||||
| AGW27560 ALH24946 EQK97643 | 100% 99.4% 100% | Q-to-K, H-to-Y | A1 | A | A B C | U3N942 A0A0N9QMM1 T5A511 | ||
| ALH24992 ALH24995 | 99.7% | A-to-T | A2 | A | D E | A0A0N9R5B3 A0A0N7G849 | ||
| T-to-S | ||||||||
| ALH25047 ALH24948 | 99.4% | S-to-L | A3 | A | F G | A0A0N9R4V2 A0A0N9QUF3 | ||
| ALH25043 | 98.9% | R-to-I, P-to-T, T-to-I, G-to-A, | B | H | A0A0N9QMS9 | |||
| ALH25054 | 99.4% | I-to-L | A-to-V | C | I | A0A0N7G845 | ||
| ALH25005 | 99.2% | H-to-Y | P-to-H | D1 | D | J | A0A0N9QUK2 | |
| ALH24999 | 98.1% | S-to-T, I-to-V, H-to-Y | A-to-V, A-to-T | D2 | D | K | A0A0N9QMT4 | |
| ALH25008 | 99.7% | Y-to-H | E1 | E | L | A0A0N9QMR3 | ||
| ALH25034 | 99.4% | E-to-K, Y-to-H | E2 | E | M | A0A0N9QMS4 | ||
| ALH25003 | 99.2% | E-to-K, Y-to-H | S-to-G | E3 | E | N | A0A0N7G850 | |
| ALH25001 | 98.4% | S-to-T, V-to-I, E-to-K, Y-to-H | A-to-V, A-to-T | E4 | E | O | A0A0N9R4Q4 | |
| Accession Number | % Similarity to AEH27625 | Amino Acid Residue Substitution | Bayesian Cluster | 3D Structure Model | AlphaFold UniProt Code | |||
|---|---|---|---|---|---|---|---|---|
| Conservative | Nonconservative | Branch | Cluster | |||||
| AEH27625 EQL04085 | 100% 100% | I-1 | I | A B | D7F2E9 T5AF56 | |||
| AFX66437 ACV60391 ACV60372 | 99.6% | D-to-N | I-2 | I | C D E | V9LW10 D7F2H1 D7F2F2 | ||
| G-to-R Q-to-L | ||||||||
| AIV43040 | 97.6% | Y-to-H, M-to-I, Q-to-R, S-to-T | T-to-I, A-to-G | II-1 | II | F | A0A0A0RCF5 | |
| ACV60417 | 97.6% | Y-to-H, M-to-I, S-to-T | T-to-I, T-to-A, A-to-G | II-2 | II | G | D7F2J7 | |
| ACV60415 | 98.8% | Y-to-H, M-to-I | T-to-G | III | H | D7F2F5 | ||
| AFX66476 | 98.8% | Y-to-H | D-to-G, V-to-A | IV-1 | IV | I | V9LWC9 | |
| AFX66472 | 97.6% | Y-to-H, D-to-N, S-to-T | D-to-G, V-to-A, A-to-T | IV-2 | IV | J | V9LVS8 | |
| ACV60363 | 99.6% | Y-to-H | V-1 | V | K | D7F2E3 | ||
| ACV60385 | 99.2% | I-to-V, Y-to-H | V-2 | V | L | D7F2G5 | ||
| ACV60399 | 99.2% | Y-to-H, Q-to-R | V-2 | V | M | D7F2H9 | ||
| AFX66401 | 99.2% | Y-to-H | D-to-I | V-2 | V | N | V9LW71 | |
| AFX66475 | 99.2% | Y-to-H | I-to-T | V-2 | V | O | V9LVU8 | |
| AFX66484 | 99.2% | Y-to-H | A-to-T | V-2 | V | P | V9LWG5 | |
| AGW27537 | 99.2% | V-to-I, Y-to-H | V-2 | V | Q | U3N6V5 | ||
| H. sinensis Strain | Genome Assembly Segment | Percentage Similarity | |
|---|---|---|---|
| MAT1-1-1 (vs. EQK97643) | MAT1-2-1 (vs. AEH27625) | ||
| Co18 | ANOV01017390 (410←1519) | 99.7% | |
| ANOV01000063 (9329→10,182) | 99.6% | ||
| 1229 | LKHE01001116 (3799←4909) | 99.7% | |
| LKHE01001605 (13,860←14,713) | 99.6% | ||
| IOZ07 | JAAVMX010000001 (6,698,911→6,700,021) | 99.7% | |
| JAAVMX000000000 | ― | ||
| ZJB12195 | LWBQ00000000 | ― | |
| LWBQ01000021 (238,873←239,726) | 99.6% | ||
| CC1406-20395 | NGJJ00000000 | ― | |
| NGJJ01000619 (23,030←23,883) | 99.6% | ||
| H. sinensis Strain or Natural C. sinensis | Transcriptome or Metatranscriptome Assembly Segment | Percentage Similarity | |
|---|---|---|---|
| MAT1-1-1 (vs. EQK97643) | MAT1-2-1 (vs. AEH27625) | ||
| H. sinensis strain L0106 | GCQL00000000 | ― | |
| GCQL01020543 (397←1143) | 99.6% | ||
| Mature natural C. sinensis (Collected at Deqin, Yunnan) | OSIN7648 (1→1065) | 94.9% | |
| OSIN7649 (1→397) | 100% | ||
| Natural C. sinensis * (Collected at Kangding, Sichuan) | GAGW01008880 (300←1127) | 100% | |
| GAGW00000000 | ― | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-Z.; Li, Y.-L.; Zhu, J.-S. Three-Dimensional Structural Heteromorphs of Mating-Type Proteins in Hirsutella sinensis and the Natural Cordyceps sinensis Insect–Fungal Complex. J. Fungi 2025, 11, 244. https://doi.org/10.3390/jof11040244
Li X-Z, Li Y-L, Zhu J-S. Three-Dimensional Structural Heteromorphs of Mating-Type Proteins in Hirsutella sinensis and the Natural Cordyceps sinensis Insect–Fungal Complex. Journal of Fungi. 2025; 11(4):244. https://doi.org/10.3390/jof11040244
Chicago/Turabian StyleLi, Xiu-Zhang, Yu-Ling Li, and Jia-Shi Zhu. 2025. "Three-Dimensional Structural Heteromorphs of Mating-Type Proteins in Hirsutella sinensis and the Natural Cordyceps sinensis Insect–Fungal Complex" Journal of Fungi 11, no. 4: 244. https://doi.org/10.3390/jof11040244
APA StyleLi, X.-Z., Li, Y.-L., & Zhu, J.-S. (2025). Three-Dimensional Structural Heteromorphs of Mating-Type Proteins in Hirsutella sinensis and the Natural Cordyceps sinensis Insect–Fungal Complex. Journal of Fungi, 11(4), 244. https://doi.org/10.3390/jof11040244