
Whole-Genome DNA Methylation Analysis of Inoculation with Trichothecium roseum in Harvested Muskmelons

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
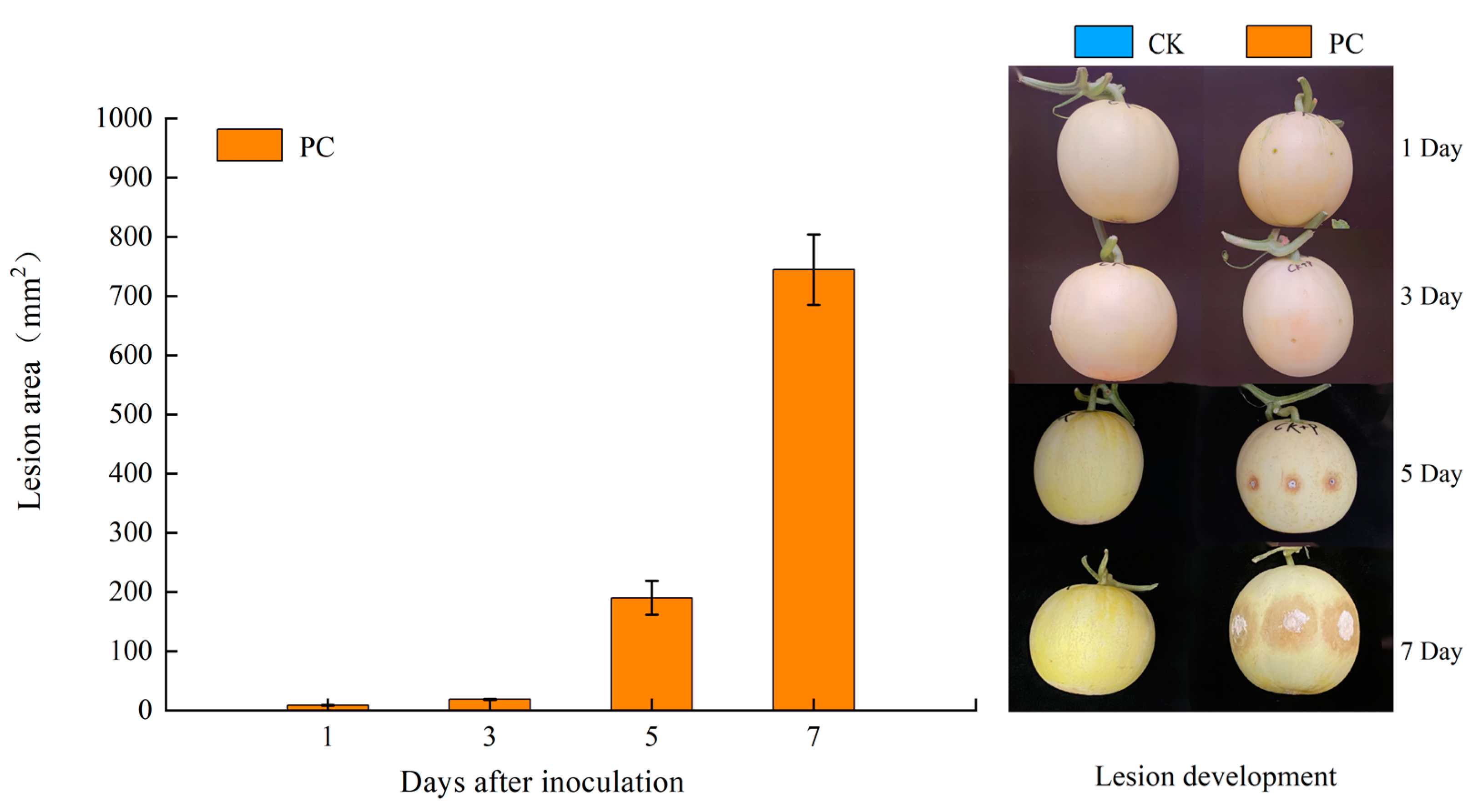
2.1. Fruit Material and Trichothecium roseum Inoculation
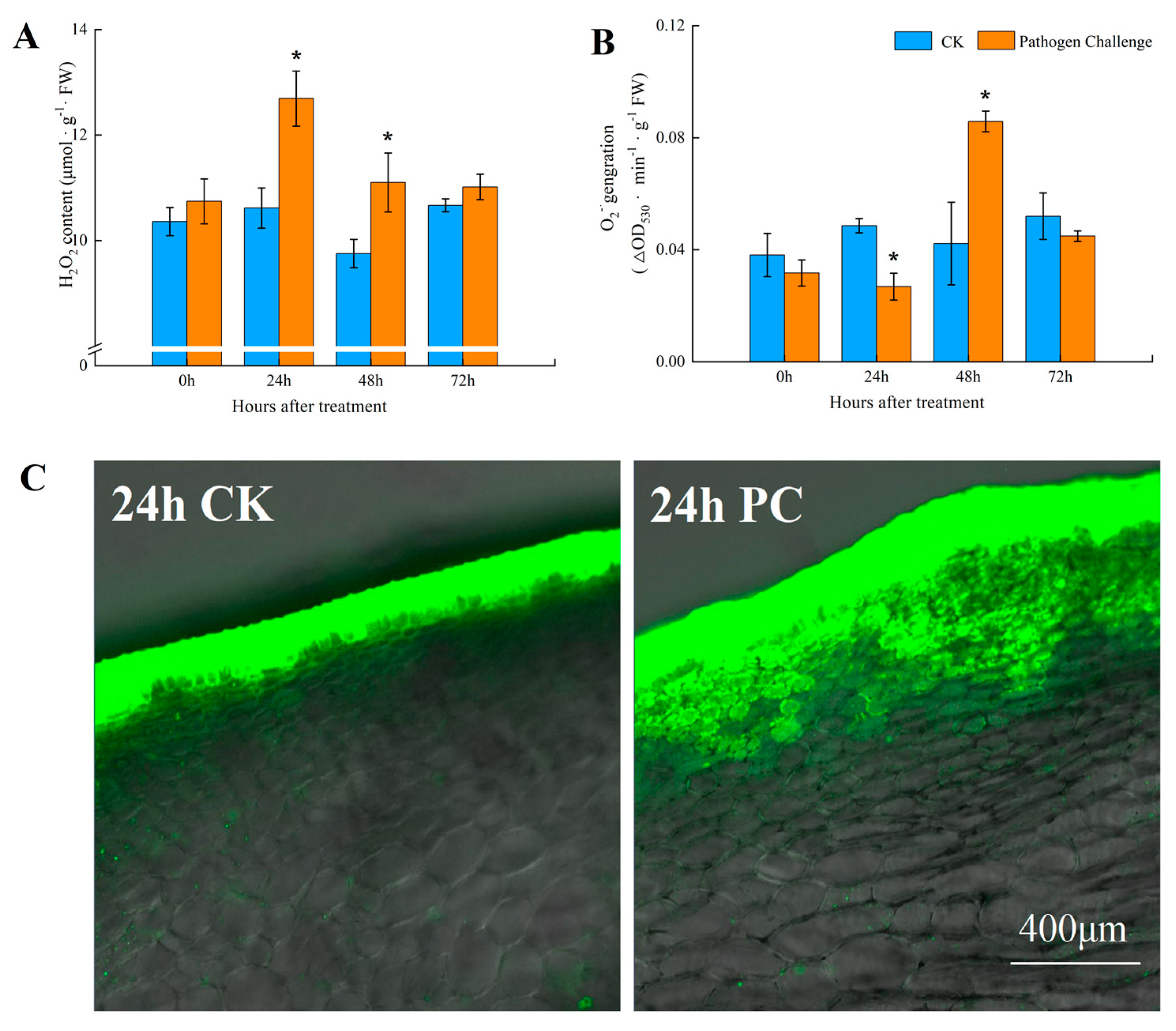
2.2. Assay and Localization for ROS Production
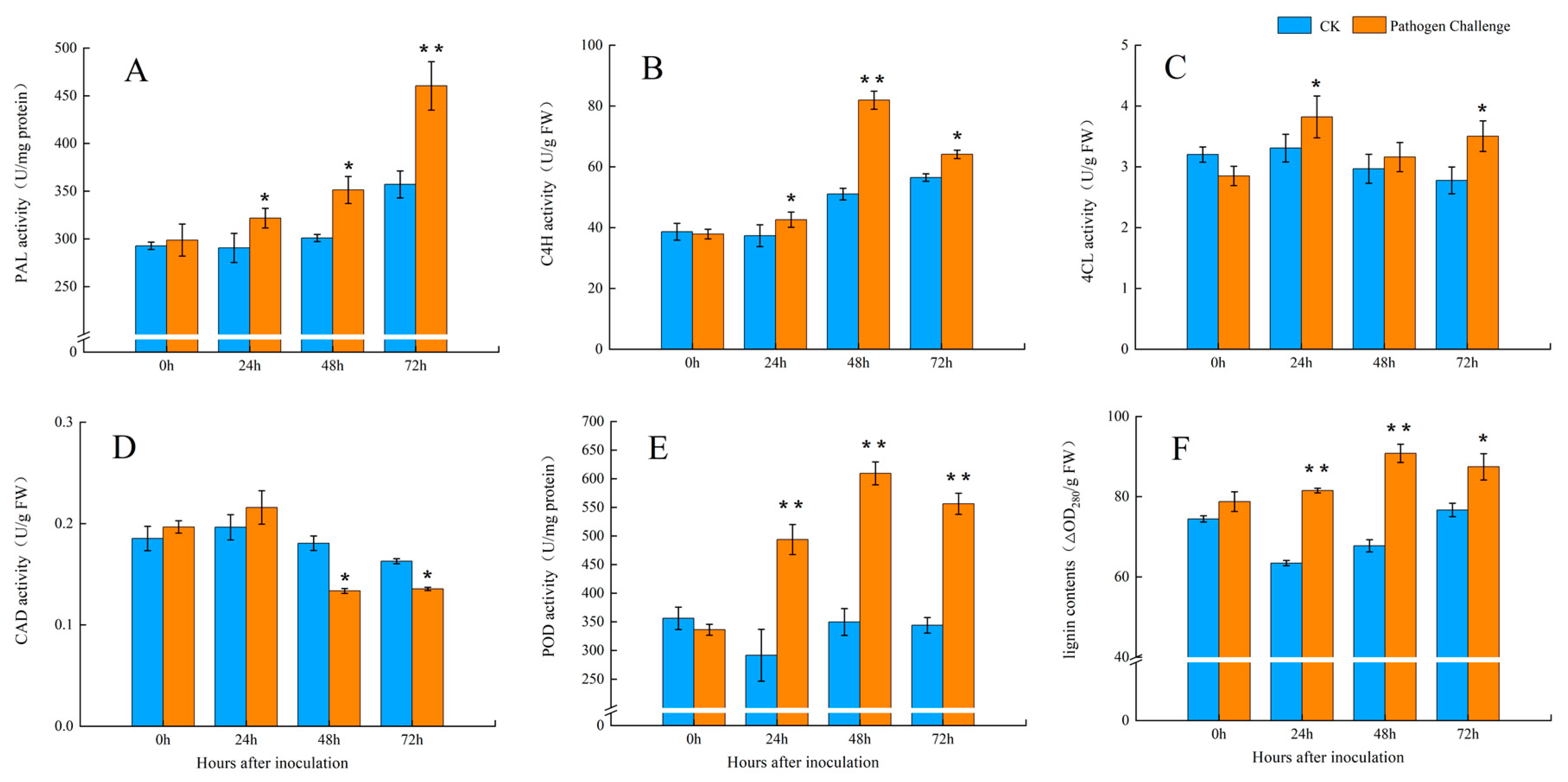
2.3. Assays for Enzyme Activity and Lignin Content
2.4. Construction of Methyl-Seq Libraries
2.5. Data Processing
2.6. RNA Sequencing and Data Analysis
2.7. Statistics Analysis
3. Results
3.1. Fungal Infection Promoted ROS Accumulation in Muskmelon Fruits
3.2. The Activities of the Key Enzymes of the Phenylpropanoid Pathway After Fungal Infection
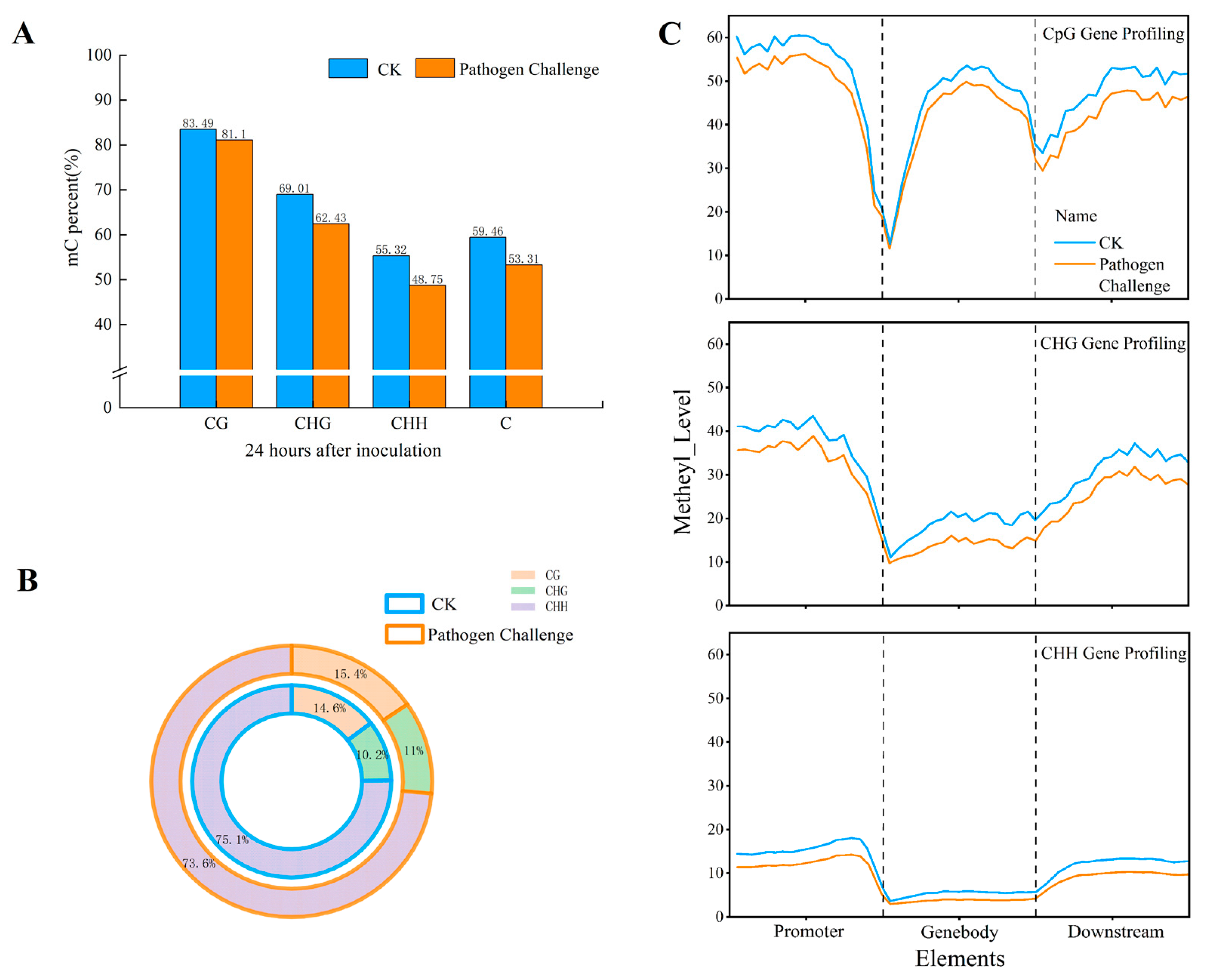
3.3. DNA Methylation Profiling
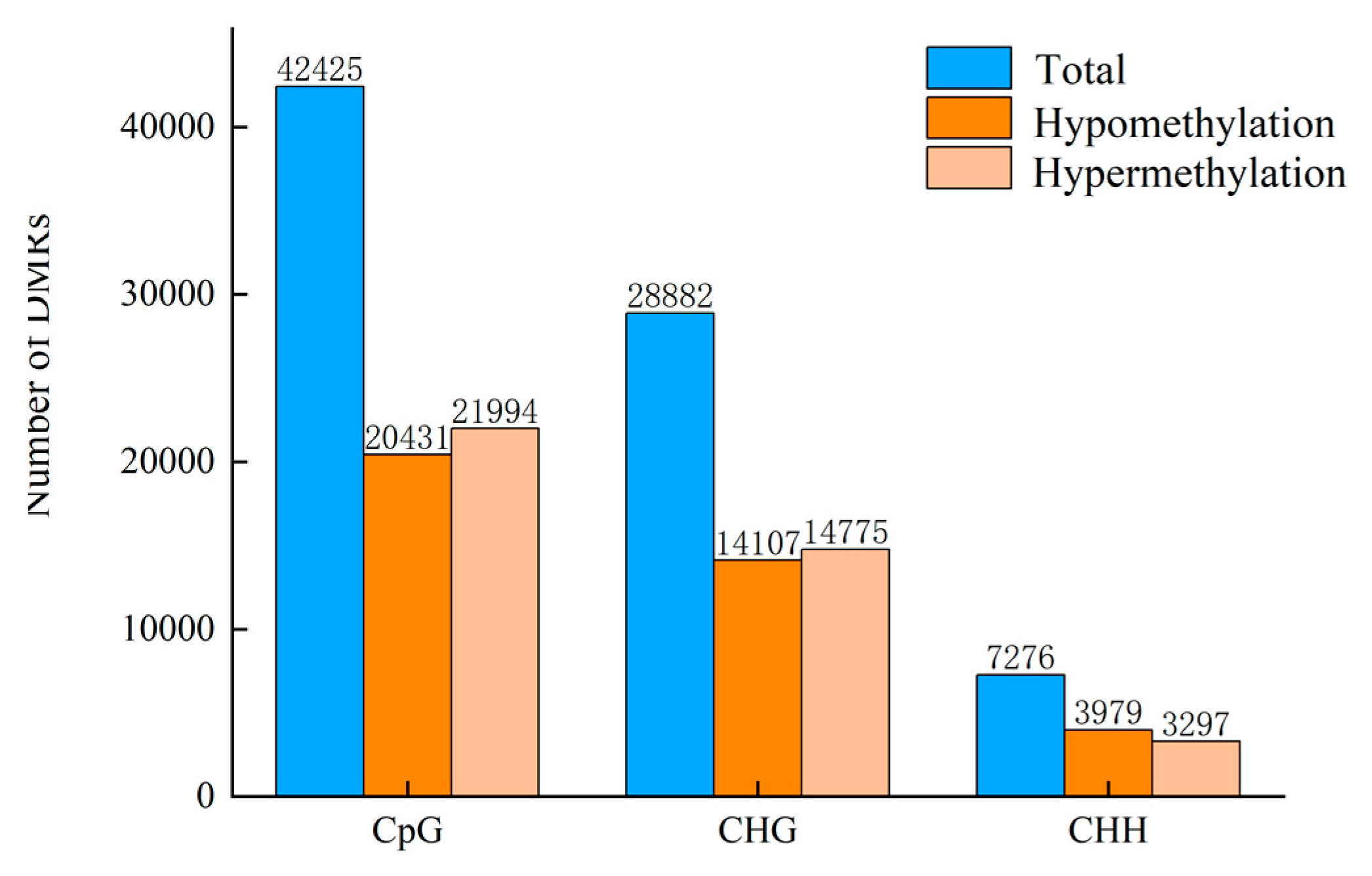
3.4. Detection of Differentially Methylated Regions
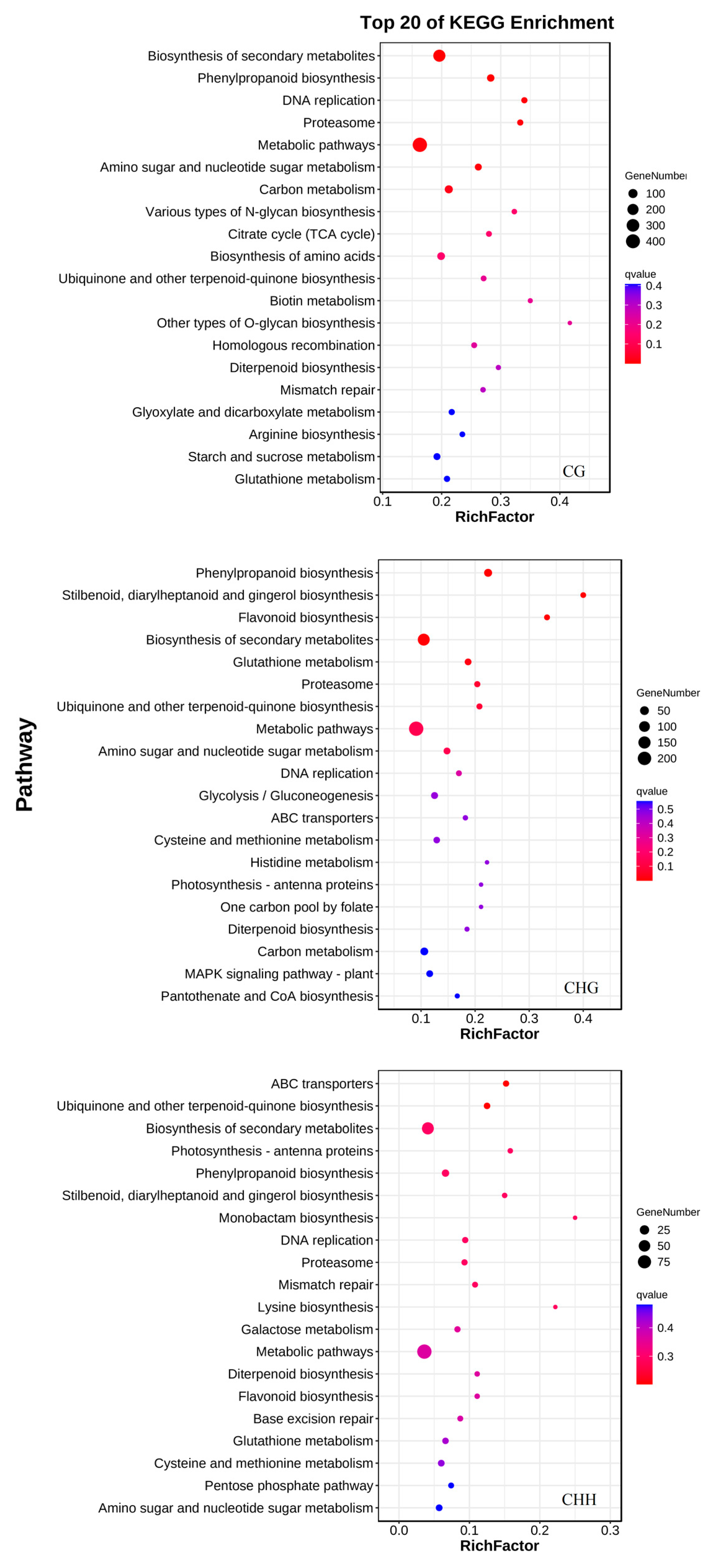
3.4.1. Gene Ontology (GO) Enrichment of Genes Associated with DMRs
3.4.2. DMR-Associated Genes Involved in Early Defense Response During T. roseum Infection
3.4.3. DMGs Involved in ROS Metabolic and Response to Oxidative Process During T. roseum Infection
3.4.4. DMGs Involved in Phenylpropanoid Metabolism During T. roseum Infection
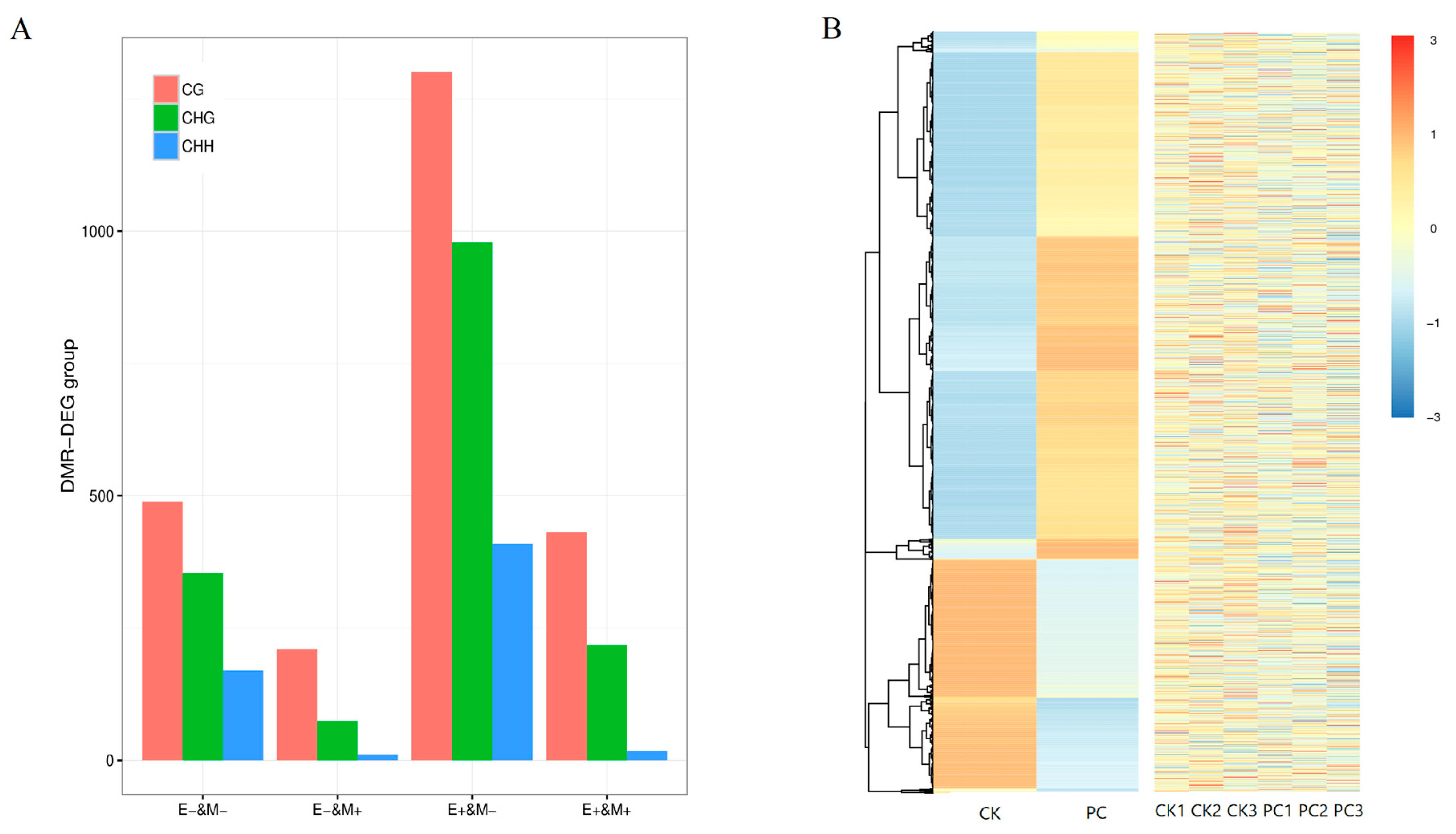
3.5. Association Between DNA Methylation and Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arora, H.; Singh, R.K.; Sharma, S.; Sharma, N.; Panchal, A.; Das, T.; Prasad, A.; Prasad, M. DNA methylation dynamics in response to abiotic and pathogen stress in plants. Plant Cell Rep. 2022, 41, 1931–1944. [Google Scholar]
- Liu, Y.; Liu, Q.; Tang, Y.; Ding, W. NtPR1a regulates resistance to Ralstonia solanacearum in Nicotiana tabacum via activating the defense-related genes. Biochem. Biophys. Res. Commun. 2019, 508, 940–945. [Google Scholar] [CrossRef]
- Zhu, W.; Hu, B.; Becker, C.; Doğan, E.S.; Berendzen, K.W.; Weigel, D.; Liu, C. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 2017, 18, 1–16. [Google Scholar]
- Bhat, R.S.; Rockey, J.; Shirasawa, K.; Tilak, I.S.; Brijesh Patil, M.P.; Reddy Lachagari, V.B. DNA methylation and expression analyses reveal epialleles for the foliar disease resistance genes in peanut (Arachis hypogaea L.). BMC Res. Notes 2020, 13, 1–7. [Google Scholar]
- Sun, Y.; Fan, M.; He, Y. DNA methylation analysis of the Citrullus lanatus response to cucumber green mottle mosaic virus infection by whole-genome bisulfite sequencing. Genes 2019, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Vishal, P.; Kaul, S.; Dhar, M.K. Epiallelic changes in known stress-responsive genes under extreme drought conditions in Brassica juncea (L.) Czern. Plant Cell Rep. 2017, 36, 203–217. [Google Scholar] [PubMed]
- Shen, S.; Wang, Z.; Shan, X.; Wang, H.; Li, L.; Lin, X.; Long, L.; Weng, K.; Liu, B.; Zou, G. Alterations in DNA methylation and genome structure in two rice mutant lines induced by high pressure. Sci. China C Life Sci. 2006, 49, 97–104. [Google Scholar]
- Shi, J.; Lu, W.; Sun, Y. Comparison of space flight and heavy ion radiation induced genomic/epigenomic mutations in rice (Oryza sativa). Life Sci. Space Res. 2014, 1, 74–79. [Google Scholar]
- Ma, X.; Wang, Q.; Wang, Y.; Ma, J.; Wu, N.; Ni, S.; Luo, T.; Zhuang, L.; Chu, C.; Cho, S.; et al. Chromosome aberrations induced by zebularine in triticale. Genome 2016, 59, 485–492. [Google Scholar] [CrossRef]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar]
- Levy, A.A.; Feldman, M. Genetic and epigenetic reprogramming of the wheat genome upon allopolyploidization. Biol. J. Linn. Soc. 2004, 82, 607–613. [Google Scholar]
- Zhao, X.; Chai, Y.; Liu, B. Epigenetic inheritance and variation of DNA methylation level and pattern in maize intra-specific hybrids. Plant Sci. 2007, 172, 930–938. [Google Scholar] [CrossRef]
- Kour, G.; Kour, B.; Kaul, S.; Dhar, M.K. Genetic and epigenetic instability of amplification-prone sequences of a novel B chromosome induced by tissue culture in Plantago lagopus L. Plant Cell Rep. 2009, 28, 1857–1867. [Google Scholar]
- Wang, C.; Wang, C.; Xu, W.; Zou, J.; Qiu, Y.; Kong, J.; Yang, Y.; Zhang, B.; Zhu, S. Epigenetic changes in the regulation of Nicotiana tabacum response to cucumber mosaic virus infection and symptom recovery through single-base resolution methylomes. Viruses 2018, 10, 402. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Chang, Y.; Lu, J.; Wu, X.; Liu, Q.; Zhang, W.; Su, X.; Zhang, B. Global methylomic and transcriptomic analyses reveal the broad participation of DNA methylation in daily gene expression regulation of Populus trichocarpa. Front. Plant Sci. 2019, 10, 243. [Google Scholar]
- Bi, Y.; Li, Y.; Ge, Y. Induced resistance in postharvest fruits and vegetables by chemicals and its mechanism. Stewart Postharvest Rev. 2007, 3, 1–7. [Google Scholar]
- Bi, Y.; Tian, S.; Guo, Y.; Ge, Y.; Qin, G. Sodium silicate reduces postharvest decay on Hami melons: Induced resistance and fungistatic effects. Plant Dis. 2006, 90, 279–283. [Google Scholar]
- Ge, Y.; Deng, H.; Bi, Y.; Li, C.; Liu, Y.; Dong, B. Postharvest ASM dipping and DPI pre-treatment regulated reactive oxygen species metabolism in muskmelon (Cucumis melo L.) fruit. Postharvest Biol. Technol. 2015, 99, 160–167. [Google Scholar]
- Liu, Y.; Ge, Y.; Bi, Y.; Li, C.; Deng, H.; Hu, L.; Dong, B. Effect of postharvest acibenzolar-S-methyl dipping on phenylpropanoid pathway metabolism in muskmelon (Cucumis melo L.) fruits. Sci. Hortic. 2014, 168, 113–119. [Google Scholar]
- Lyu, L.; Bi, Y.; Li, S.; Xue, H.; Zhang, Z.; Prusky, D.B. Early defense responses involved in mitochondrial energy metabolism and reactive oxygen species accumulation in harvested muskmelons infected by Trichothecium roseum. J. Agric. Food Chem. 2019, 67, 4337–4345. [Google Scholar]
- Lyu, L.; Bi, Y.; Li, S.; Xue, H.; Li, Y.; Prusky, D.B. Sodium silicate prime defense responses in harvested muskmelon by regulating mitochondrial energy metabolism and reactive oxygen species production. Food Chem. 2019, 289, 369–376. [Google Scholar] [CrossRef]
- Li, W.; Bi, Y.; Ge, Y.; Li, Y.; Wang, J.; Wang, Y. Effects of postharvest sodium silicate treatment on pink rot disease and oxidative stress-antioxidative system in muskmelon fruit. Eur. Food Res. Technol. 2012, 234, 137–145. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, Y.; Bi, Y.; Ge, Y.; Wang, Y.; Fan, C.; Li, D.; Deng, H. Postharvest BTH treatment induced disease resistance and enhanced reactive oxygen species metabolism in muskmelon (Cucumis melo L.) fruit. Eur. Food Res. Technol. 2012, 234, 963–971. [Google Scholar] [CrossRef]
- Elstner, E.F.; Heupel, A. Inhibition of nitrite formation from hydroxylammoniumchloride: A simple assay for superoxide dismutase. Anal. Biochem. 1976, 70, 616–620. [Google Scholar] [CrossRef]
- Patterson, B.D.; MacRae, E.A.; Ferguson, I.B. Estimation of hydrogen peroxide in plant extracts using titanium (IV). Anal. Biochem. 1984, 139, 487–492. [Google Scholar] [CrossRef]
- Lamb, C.J.; Rubery, P.H. A spectrophotometric assay for trans-cinnamic acid 4-hydroxylase activity. Anal. Biochem. 1975, 68, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Voo, K.S.; Whetten, R.W.; O'Malley, D.M.; Sederoff, R.R. 4-Coumarate: Coenzyme A ligase from loblolly pine xylem (isolation, characterization, and complementary DNA cloning). Plant Physiol. 1995, 108, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Goffner, D.; Joffroy, I.; Grima-Pettenati, J.; Halpin, C.; Knight, M.E.; Schuch, W.T.; Boudet, A.M. Purification and characterization of isoforms of cinnamyl alcohol dehydrogenase from Eucalyptus Xylem. Planta 1992, 188, 48–53. [Google Scholar] [CrossRef]
- Venisse, J.S.; Gullner, G.; Brisset, M.N. Evidence for the involvement of an oxidative stress in the initiation of infection of pear by Erwinia amylovora. Plant Physiol. 2001, 125, 2164–2172. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [PubMed]
- Wagih, O. ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 2017, 33, 3645–3647. [Google Scholar]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, T.; Dong, X.; Li, D.Z.; Liu, A. Genomic DNA methylation analyses reveal the distinct profiles in castor bean seeds with persistent endosperms. Plant Physiol. 2016, 171, 1242–1258. [Google Scholar] [CrossRef]
- Torres, M.A. ROS in biotic interactions. Physiol. Plant. 2010, 138, 414–429. [Google Scholar]
- Xiao, X.O.; Lin, W.; Li, K.; Li, W.; Gao, X.; Lv, L. Early burst of reactive oxygen species positively regulates resistance of eggplant against bacterial wilt. J. Phytopathol. 2017, 165, 652–661. [Google Scholar]
- Huang, S.; Van Aken, O.; Schwarzländer, M.; Belt, K.; Millar, A.H. The roles of mitochondrial reactive oxygen species in cellular signaling and stress response in plants. Plant Physiol. 2016, 171, 1551–1559. [Google Scholar]
- Liu, J.; He, Z. Small DNA methylation, big player in plant abiotic stress responses and memory. Front. Plant Sci. 2020, 11, 595603. [Google Scholar]
- Villagómez-Aranda, A.L.; García-Ortega, L.F.; Torres-Pacheco, I.; Guevara-González, R.G. Whole-Genome DNA methylation analysis in hydrogen peroxide overproducing transgenic tobacco resistant to biotic and abiotic stresses. Plants 2021, 10, 178. [Google Scholar] [CrossRef]
- Bi, Y.; Ge, Y.H.; Li, Y.C.; Wang, J.J.; Miao, X.Y.; Li, X.W. Postharvest acibenzolar-S-methyl treatment suppresses decay and induces resistance in Hami melons. In Proceedings of the IV International Conference on Managing Quality in Chains-The Integrated View on Fruits and Vegetables Quality, Bangkok, Thailand, 30 June 2006; Volume 712, pp. 393–400. [Google Scholar]
- Ge, Y.; Bi, Y.; Li, Y.; Wang, Y. Resistance of harvested fruits and vegetables to diseases induced by ASM and its mechanism. Sci. Agric. Sin. 2012, 45, 3357–3362. [Google Scholar]
- Hématy, K.; Cherk, C.; Somerville, S. Host–pathogen warfare at the plant cell wall. Curr. Opin. Plant Biol. 2009, 12, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, X.; Wu, Z.; An, H.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Tu, J. Genome-wide DNA methylation comparison between Brassica napus genic male sterile line and restorer line. Int. J. Mol. Sci. 2018, 19, 2689. [Google Scholar] [CrossRef] [PubMed]
- López Sánchez, A.; Stassen, J.H.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de) methylation in immune responsiveness of Arabidopsis. Plant J. 2016, 88, 361–374. [Google Scholar]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 1–15. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar]
- Pavet, V.; Quintero, C.; Cecchini, N.M.; Rosa, A.L.; Alvarez, M.E. Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol. Plant Microbe Interact. 2006, 19, 577–587. [Google Scholar]
- Alonso, C.; Ramos-Cruz, D.; Becker, C. The role of plant epigenetics in biotic interactions. New Phytol. 2019, 221, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Kumar, G.S.; Khan, M.; Doohan, F.M. Brassinosteroid enhances resistance to fusarium diseases of barley. Phytopathology 2013, 103, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, W.; Yang, X.; Zhang, J.; Shi, Q. Comparative methylome reveals regulatory roles of DNA methylation in melon resistance to Podosphaera xanthii. Plant Sci. 2021, 309, 110954. [Google Scholar] [CrossRef]
- Viggiano, L.; de Pinto, M.C. Dynamic DNA methylation patterns in stress response. In Plant Epigenetics; Springer: Cham, Switzerland, 2017; pp. 281–302. [Google Scholar]
- He, C.; Zhang, H.Y.; Zhang, Y.X.; Fu, P.; You, L.L.; Xiao, W.B.; Liao, J.L. Cytosine methylations in the promoter regions of genes involved in the cellular oxidation equilibrium pathways affect rice heat tolerance. BMC Genom. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Xie, H.; Zheng, Y.; Xue, M.; Huang, Y.; Qian, D.; Zhao, M.; Li, J. DNA methylation-mediated ROS production contributes to seed abortion in litchi. Mol. Hortic. 2024, 4, 12. [Google Scholar] [CrossRef]
- Huang, J.; Gu, M.; Lai, Z.; Fan, B.; Shi, K.; Zhou, Y.H.; Yu, J.Q.; Chen, Z. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiol. 2010, 153, 1526–1538. [Google Scholar] [CrossRef]
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From biosynthesis to health benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef]
- Vega-Muñoz, I.; Feregrino-Pérez, A.A.; Torres-Pacheco, I.; Guevara-González, R.G. Exogenous fragmented DNA acts as a damage-associated molecular pattern (DAMP) inducing changes in CpG DNA methylation and defence-related responses in Lactuca sativa. Funct. Plant Biol. 2018, 45, 1065–1072. [Google Scholar] [CrossRef]
- Vanholme, R.; Cesarino, I.; Rataj, K.; Xiao, Y.; Sundin, L.; Goeminne, G.; Kim, H.; Cross, J.; Boerjan, W. Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 2013, 341, 1103–1106. [Google Scholar] [CrossRef]
- Zhao, Z.; Dong, Y.; Wang, J.; Zhang, G.; Zhang, Z.; Zhang, A. Comparative transcriptome analysis of melon (Cucumis melo L.) reveals candidate genes and pathways involved in powdery resistance. Sci. Rep. 2022, 12, 4396. [Google Scholar]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar]
- Gao, S.; Ma, W.; Lyu, X.; Cao, X.; Yao, Y. Melatonin may increase disease resistance and flavonoid biosynthesis through effects on DNA methylation and gene expression in grape berries. BMC Plant Biol. 2020, 20, 1–15. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PC vs. CK 24 h Comparison | Total DMRs | Hyper DMRs | Hypo DMRs | Exon Hyper | Exon Hypo | Intron Hyper | Intron Hypo | Promoter Hyper | Promoter Hypo | Downstream Hyper | Downstream Hypo | Intergenic Hyper | Intergenic Hypo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CpG | 42,425 | 21,994 | 20,431 | 9359 | 8973 | 9685 | 9080 | 4065 | 4165 | 4809 | 4543 | 7219 | 6451 |
| CHG | 28,882 | 14,775 | 14,107 | 1809 | 2076 | 2342 | 2907 | 2539 | 2617 | 1889 | 1858 | 8297 | 7383 |
| CHH | 7276 | 3297 | 3979 | 516 | 788 | 442 | 656 | 903 | 1068 | 608 | 651 | 1499 | 1802 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, L.; Li, L.; Zhao, C.; Ning, Y.; Luo, Y.; He, X.; Nan, M. Whole-Genome DNA Methylation Analysis of Inoculation with Trichothecium roseum in Harvested Muskmelons. J. Fungi 2025, 11, 243. https://doi.org/10.3390/jof11040243
Lyu L, Li L, Zhao C, Ning Y, Luo Y, He X, Nan M. Whole-Genome DNA Methylation Analysis of Inoculation with Trichothecium roseum in Harvested Muskmelons. Journal of Fungi. 2025; 11(4):243. https://doi.org/10.3390/jof11040243
Chicago/Turabian StyleLyu, Liang, Lei Li, Chenglong Zhao, Yuchao Ning, Yawen Luo, Xining He, and Mina Nan. 2025. "Whole-Genome DNA Methylation Analysis of Inoculation with Trichothecium roseum in Harvested Muskmelons" Journal of Fungi 11, no. 4: 243. https://doi.org/10.3390/jof11040243
APA StyleLyu, L., Li, L., Zhao, C., Ning, Y., Luo, Y., He, X., & Nan, M. (2025). Whole-Genome DNA Methylation Analysis of Inoculation with Trichothecium roseum in Harvested Muskmelons. Journal of Fungi, 11(4), 243. https://doi.org/10.3390/jof11040243