
Whole-Genome Sequencing and Fine Map Analysis of Pholiota nameko


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions of the Samples
2.2. Fruiting Body Tissue Culture and Mycelium Collection
2.3. Genomic DNA Extraction
2.4. Genome Sequencing and Raw Data Preprocessing
2.5. Nanopore Sequencing Data Processing
2.6. Genome Splicing and Completion
2.6.1. Gene Component Analysis
2.6.2. Gene Annotation
2.7. Determination of Mating Site
2.8. Identification of CAZymes
2.9. Pan-Genome Analysis
3. Results
3.1. Cultivation and Strain Isolation
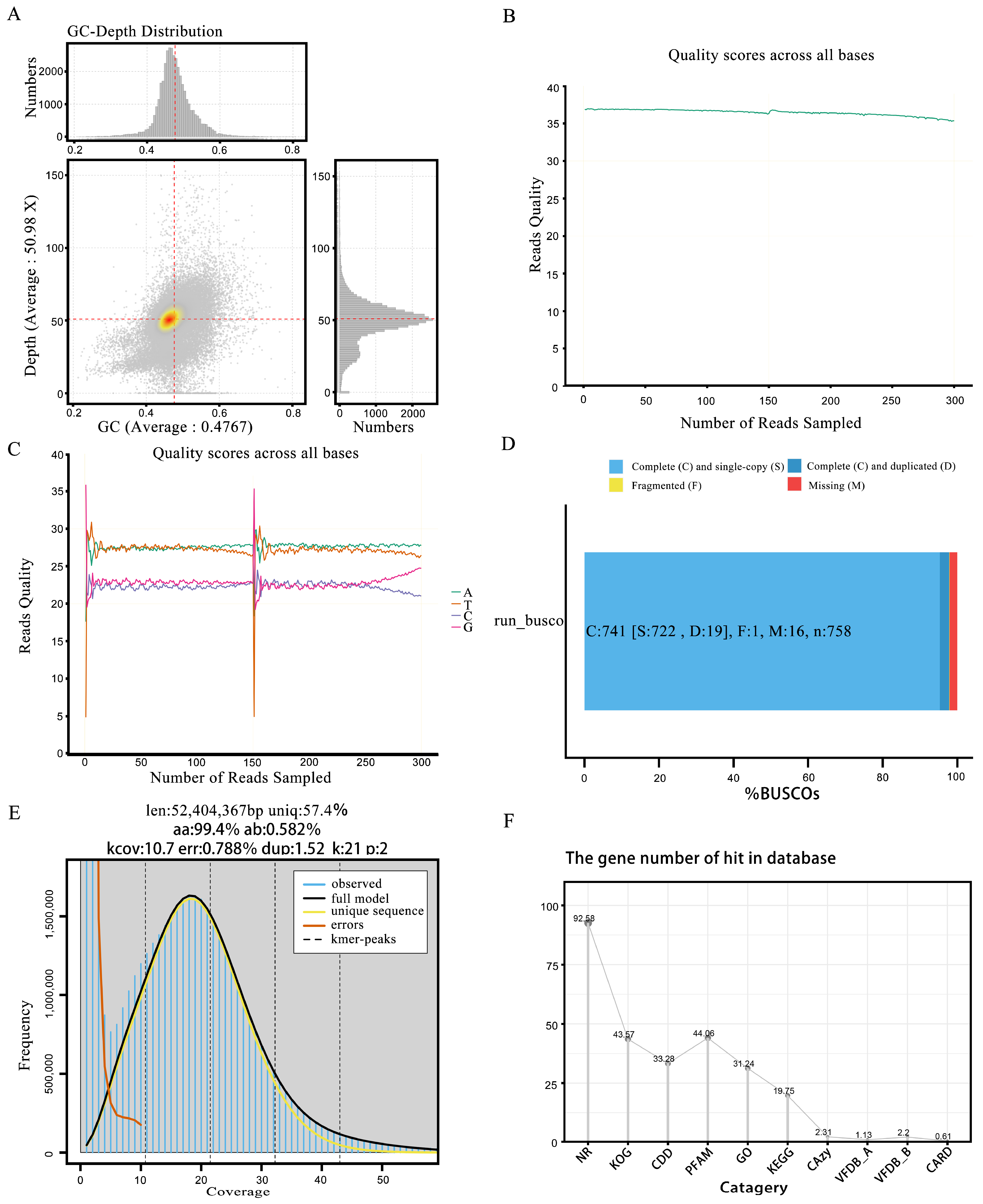
3.2. Genome Sequence Assembly
3.3. Genomic Analysis
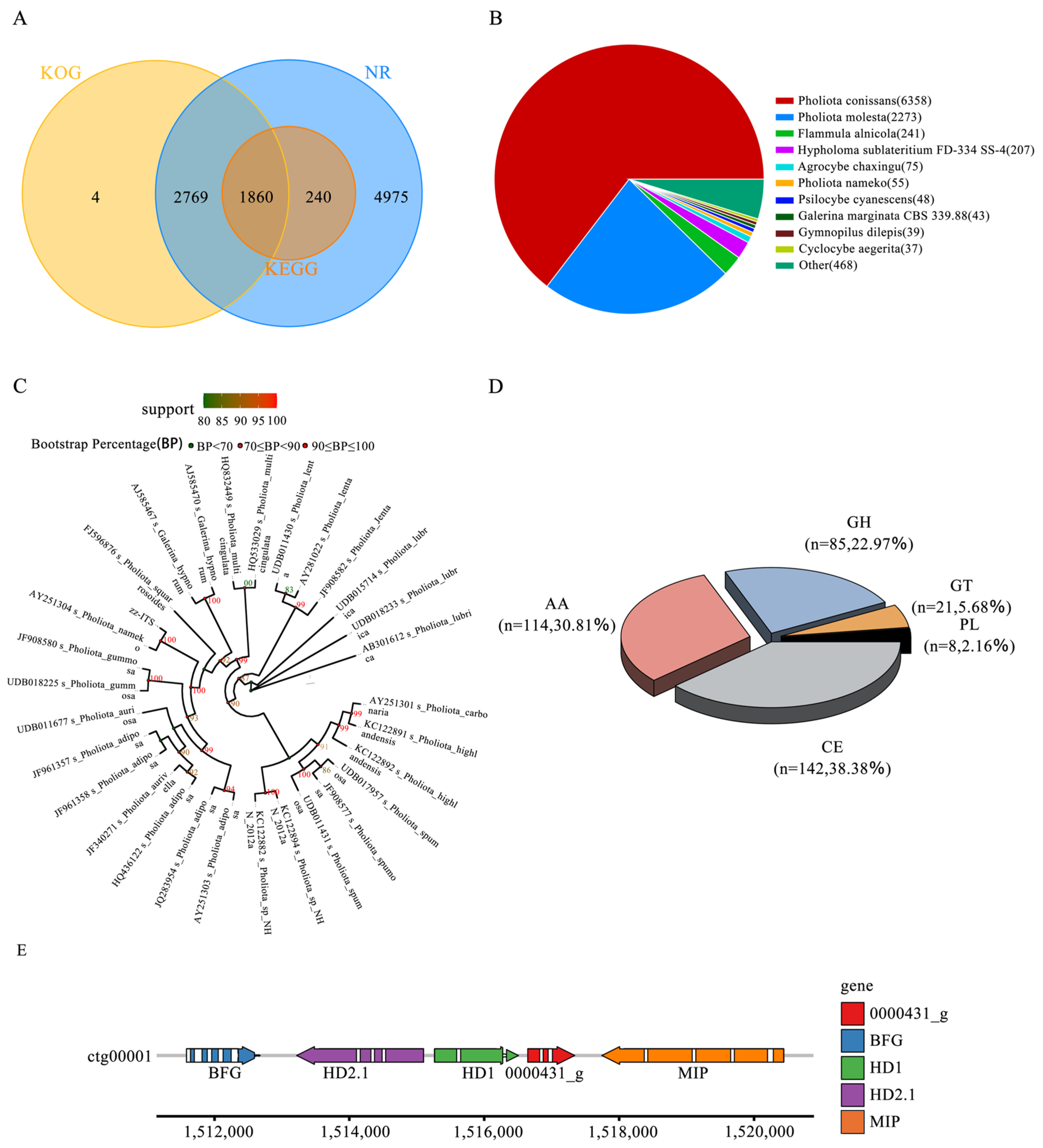
3.4. Phylogenetic Analysis
3.5. CAZymes Analysis
3.6. Identification of Mating-Type Sites
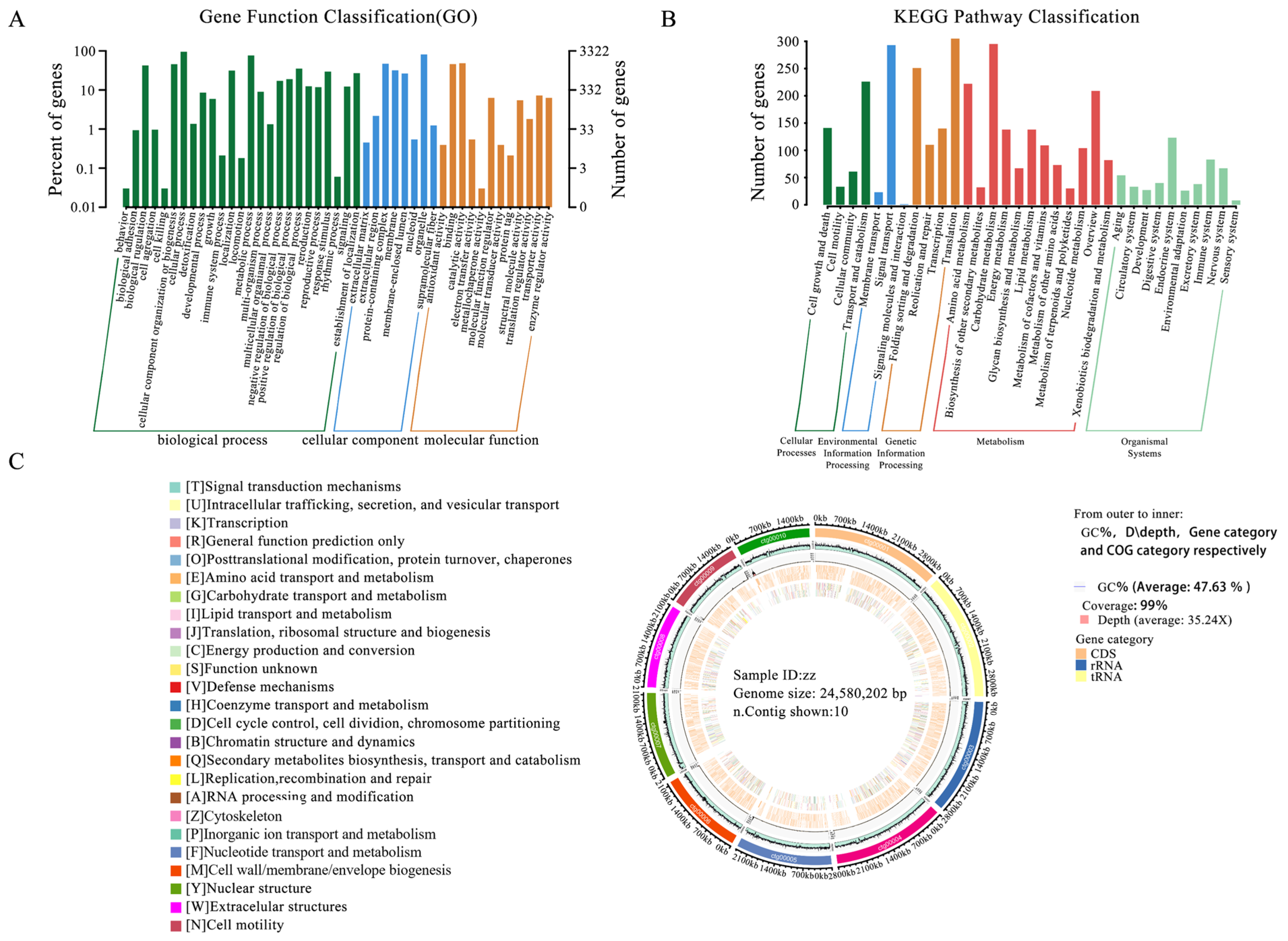
3.7. Genome Functional Annotation
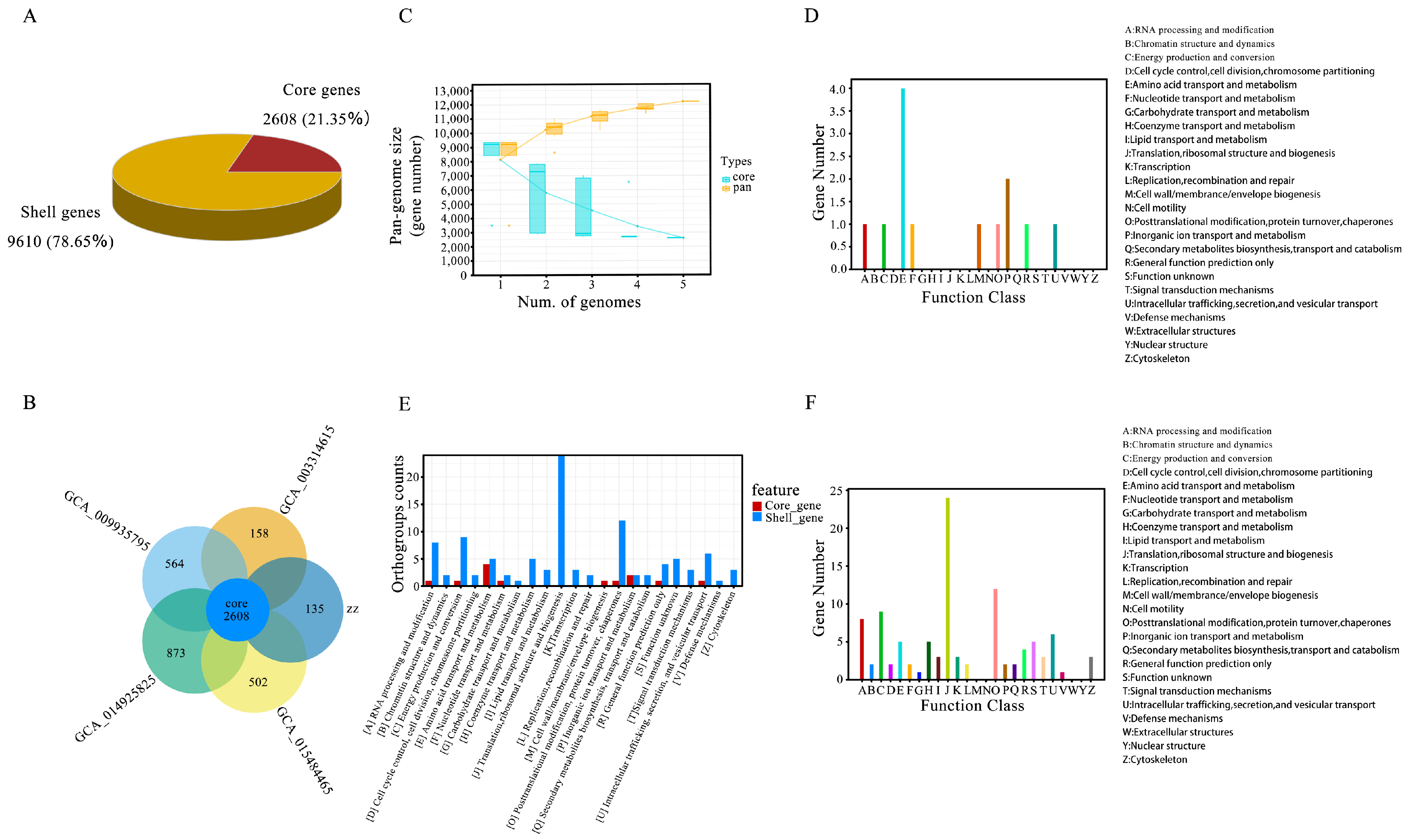
3.8. Pan-Genome Analysis Annotation
3.9. Virulence Factors and Resistance Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adhikari, M.K.; Watanabe, K.; Parajuli, G. A new variety of Pholiota microspora (Agaricales) from Nepal. Biodivers. J. Biol. Divers. 2014, 15, 101–103. [Google Scholar]
- Meng, L.; Fu, Y.; Li, D.; Sun, X.; Chen, Y.; Li, X.; Xu, S.; Li, X.; Li, C.; Song, B. Effects of corn stalk cultivation substrate on the growth of the slippery mushroom (Pholiota microspora). RSC Adv. 2019, 9, 5347–5353. [Google Scholar] [CrossRef]
- Hirao, A.S.; Kumata, A.; Takagi, T.; Sasaki, Y.; Shigihara, T.; Kimura, E.; Kaneko, S. Japanese “nameko” mushrooms (Pholiota microspora) produced via sawdust-based cultivation exhibit severe genetic bottleneck associated with a single founder. Mycoscience 2022, 63, 79–87. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, W.; Shen, Y.; Wang, Y.; Dai, Y.-C. Edible mushroom cultivation for food security and rural development in China: Bio-innovation, technological dissemination and marketing. Sustainability 2014, 6, 2961–2973. [Google Scholar] [CrossRef]
- Singh, P.; Varshney, V.K. Nutritional Attributes and Nonvolatile Taste Components of Medicinally Important Wild False Earthstar Mushroom, Astraeus hygrometricus (Agaricomycetes), from India. Int. J. Med. Mushrooms 2020, 22, 909–918. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, B.; Yan, G.; Wu, H.; Han, Y.; Cui, H. Chemical properties and anti-fatigue effect of polysaccharide from Pholiota nameko. J. Food Biochem. 2022, 46, e14015. [Google Scholar] [CrossRef]
- Zheng, L.; Zhai, G.; Zhang, J.; Wang, L.; Ma, Z.; Jia, M.; Jia, L. Antihyperlipidemic and hepatoprotective activities of mycelia zinc polysaccharide from Pholiota nameko SW-02. Int. J. Biol. Macromol. 2014, 70, 523–529. [Google Scholar] [CrossRef]
- Lisiecka, J.; Prasad, R.; Jasinska, A. The utilisation of Pholiota nameko, Hypsizygus marmoreus, and Hericium erinaceus spent mushroom substrates in Pleurotus ostreatus cultivation. Horticulturae 2021, 7, 396. [Google Scholar] [CrossRef]
- Xie ChunQin, X.C.; Zhao GuiHua, Z.G.; Zheng GuangYao, Z.G.; Xie ZhengLin, X.Z.; Chen Yi, C.Y.; Chu ChengZhen, C.C.; Jiang YuJuan, J.Y. Cultivation matrix development study of wood rotting edible fungi using woody waste of poplar timber processing as the materials. J. West China For. Sci. 2012, 41, 73–77. [Google Scholar]
- Aimi, T.; Yoshida, R.; Ishikawa, M.; Bao, D.; Kitamoto, Y. Identification and linkage mapping of the genes for the putative homeodomain protein (hox1) and the putative pheromone receptor protein homologue (rcb1) in a bipolar basidiomycete, Pholiota nameko. Curr. Genet. 2005, 48, 184–194. [Google Scholar] [CrossRef]
- Yi, R.; Tachikawa, T.; Ishikawa, M.; Mukaiyama, H.; Bao, D.; Aimi, T. Genomic structure of the A mating-type locus in a bipolar basidiomycete, Pholiota nameko. Mycol. Res. 2009, 113, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Ahn, D.H.; Kim, S.J.; Lee, H.; Oh, T.-J.; Lee, J.E.; Park, H. Advantages of single-molecule real-time sequencing in high-GC content genomes. PLoS ONE 2013, 8, e68824. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Park, S.-G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.-S.; Ro, H.-S.; Kim, M.; Ryoo, R.; Rhee, S.-K. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Feng, X.-l.; Wang, Z.-x.; Qi, J. The first whole genome sequencing of Agaricus bitorquis and its metabolite profiling. J. Fungi 2023, 9, 485. [Google Scholar] [CrossRef]
- Li, S.; Zhao, S.; Hu, C.; Mao, C.; Guo, L.; Yu, H.; Yu, H. Whole genome sequence of an edible mushroom Stropharia rugosoannulata (Daqiugaigu). J. Fungi 2022, 8, 99. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, S.; Yang, N.; Qin, L. Whole-genome sequencing and comparative genomics analysis of the wild edible mushroom (Gomphus purpuraceus) provide insights into its potential food application and artificial domestication. Genes 2022, 13, 1628. [Google Scholar] [CrossRef]
- Sun, X.; Yang, C.; Ma, Y.; Zhang, J.; Wang, L. Research progress of Auricularia heimuer on cultivation physiology and molecular biology. Front. Microbiol. 2022, 13, 1048249. [Google Scholar] [CrossRef]
- Farr, E.R.; Miller, O.K., Jr.; Farr, D.F. Biosystematic studies in the genus Pholiota, stirps Adiposa. Can. J. Bot. 1977, 55, 1167–1180. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Ng, T.B.; Chen, Z.; Qiao, W.; Liu, F. Purification and characterization of a novel antitumor protein with antioxidant and deoxyribonuclease activity from edible mushroom Pholiota nameko. Biochimie 2014, 99, 28–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Wang, S.; Jiang, N.; Shao, L.; Chen, J. Identification of drug resistance-related virulence gene mutations in 667 clinical Mycobacterium tuberculosis isolates. J. Infect. Dev. Ctries. 2024, 18, 1404–1412. [Google Scholar] [CrossRef]
- Zhao, H.; He, Y.; Wang, Y.; He, X.; Zhao, R.; Liu, B. Analysis of microbial community evolution, autolysis phenomena, and energy metabolism pathways in Pholiota nameko endophytes. Front. Microbiol. 2024, 15, 1319886. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-H.; Kong, W.-S.; Kim, G.-H.; Jhune, C.-S.; You, C.-H.; Yoo, Y.-B.; Kim, K.-H. Analysis of cultural characteristics and phylogenic relationships of collected strains of Pholiota species. Mycobiology 2003, 31, 200–204. [Google Scholar] [CrossRef]
- Wu, J.P.; Fu, Z.Y.; Wang, X.J.; Wang, J.B. Domestication and cultivation of Pholiota squarrosa. Acta Edulis Fungi 2011, 18, 24–25. [Google Scholar]
- Caiping, L.; Baohua, P.; Yuhau, Y.; Yigong, Z. A preliminary study on the biological characteistics fo Leucopaxillus candidus mycelia. Shi Yong Jun Xue Bao Online 2004, 11, 24–29. [Google Scholar]
- Li, Q.; Chen, J.; Liu, J.; Yu, H.; Zhang, L.; Song, C.; Li, Y.; Jiang, N.; Tan, Q.; Shang, X. De novo sequencing and comparative transcriptome analyses provide first insights into polysaccharide biosynthesis during fruiting body development of Lentinula edodes. Front. Microbiol. 2021, 12, 627099. [Google Scholar] [CrossRef]
- Wang, S.X.; Liu, Y.; Xu, F.; Zhao, S.; Wang, L.Q.; Geng, X.L.; Meng, L.L. Domestication and cultivation of Oudemansiella canarii. Acta Edulis Fungi 2013, 20, 31–34. [Google Scholar]
- Nguyen, B.T.T.; Ngo, N.X.; Van Le, V.; Nguyen, L.T.; Kana, R.; Nguyen, H.D. Optimal culture conditions for mycelial growth and fruiting body formation of Ling Zhi mushroom Ganoderma lucidum strain GA3. Vietnam J. Sci. Technol. Eng. 2019, 61, 62–67. [Google Scholar] [CrossRef]
- Jin, Y.C.; Li, Y. Selected biological characteristics and artificial cultivation of Phyllotopsis nidulans. Acta Edulis Fungi 2012, 19, 35–38. [Google Scholar]
- Muraguchi, H.; Umezawa, K.; Niikura, M.; Yoshida, M.; Kozaki, T.; Ishii, K.; Sakai, K.; Shimizu, M.; Nakahori, K.; Sakamoto, Y. Strand-specific RNA-seq analyses of fruiting body development in Coprinopsis cinerea. PLoS ONE 2015, 10, e0141586. [Google Scholar] [CrossRef]
- Yoo, Y.-B.; Oh, J.A.; Oh, Y.-L.; Moon, J.; Shin, P.-G.; Jang, K.-Y.; Kong, W.-S. Characterization of cultures isolated from fruiting body tissue in Armillaria gallica. J. Mushroom 2013, 11, 63–68. [Google Scholar] [CrossRef]
- Treuner-Lange, A.; Bruckskotten, M.; Rupp, O.; Goesmann, A.; Søgaard-Andersen, L. Complete genome sequence of the fruiting myxobacterium Myxococcus macrosporus strain DSM 14697, generated by PacBio sequencing. Genome Announc. 2017, 5, e01127-17 . [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-P.; Liang, Y.; Dai, Y.-T.; Yang, C.-T.; Duan, M.-Z.; Zhang, Z.; Hu, S.-N.; Zhang, Z.-W.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Shrivastava, P.; Das, S. Principles and Practices of DNA Analysis: A Laboratory Manual for Forensic DNA Typing; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13, S8. [Google Scholar] [CrossRef]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Sf, A. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. In Genome Informatics 2009: Genome Informatics Series Volume 23; World Scientific: Singapore, 2009; pp. 205–211. [Google Scholar]
- Rahman, M.S.; Shimul, M.E.K.; Parvez, M.A.K. Comprehensive analysis of genomic variation, pan-genome and biosynthetic potential of Corynebacterium glutamicum strains. PLoS ONE 2024, 19, e0299588. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Nysten, J.; Sofras, D.; Van Dijck, P. One species, many faces: The underappreciated importance of strain diversity. PLoS Pathog. 2024, 20, e1011931. [Google Scholar] [CrossRef] [PubMed]
- Inglin, R.C.; Meile, L.; Stevens, M.J. Clustering of pan-and core-genome of Lactobacillus provides novel evolutionary insights for differentiation. BMC Genom. 2018, 19, 284. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, T.; Ferretti, P.; Maistrenko, O.M.; Bork, P. Diversity within species: Interpreting strains in microbiomes. Nat. Rev. Microbiol. 2020, 18, 491–506. [Google Scholar] [CrossRef]
- Gabor, C.E.; Hazen, T.H.; Delaine-Elias, B.C.; Rasko, D.A.; Barry, E.M. Genomic, transcriptomic, and phenotypic differences among archetype Shigella flexneri strains of serotypes 2a, 3a, and 6. Msphere 2023, 8, e00408–e00423. [Google Scholar] [CrossRef]
- Massouras, A.; Hens, K.; Gubelmann, C.; Uplekar, S.; Decouttere, F.; Rougemont, J.; Cole, S.T.; Deplancke, B. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat. Methods 2010, 7, 485–486. [Google Scholar] [CrossRef]
- Bao, D.; Gong, M.; Zheng, H.; Chen, M.; Zhang, L.; Wang, H.; Jiang, J.; Wu, L.; Zhu, Y.; Zhu, G. Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS ONE 2013, 8, e58294. [Google Scholar] [CrossRef]
- Garron, M.-L.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, M.; Sun, Y.; Li, Q.; Liu, J.; Song, C.; Shang, X.; Tan, Q.; Zhang, L.; Yu, H. Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 2022, 13, 941889. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Zheng, C.; Chitsaz, F.; Derbyshire, M.K.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Lanczycki, C.J. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2012, 41, D348–D352. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Zhou, M.; Wu, Y.; Kudinha, T.; Jia, P.; Wang, L.; Xu, Y.; Yang, Q. Comprehensive pathogen identification, antibiotic resistance, and virulence genes prediction directly from simulated blood samples and positive blood cultures by nanopore metagenomic sequencing. Front. Genet. 2021, 12, 620009. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Phillips, C.M.; Beeson IV, W.T.; Cate, J.H.; Marletta, M.A. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 2011, 6, 1399–1406. [Google Scholar] [CrossRef]
- Shi, Q.; Abdel-Hamid, A.M.; Sun, Z.; Cheng, Y.; Tu, T.; Cann, I.; Yao, B.; Zhu, W. Carbohydrate-binding modules facilitate the enzymatic hydrolysis of lignocellulosic biomass: Releasing reducing sugars and dissociative lignin available for producing biofuels and chemicals. Biotechnol. Adv. 2023, 65, 108126. [Google Scholar] [CrossRef]
- Mosbech, C.; Holck, J.; Meyer, A.; Agger, J.W. Enzyme kinetics of fungal glucuronoyl esterases on natural lignin-carbohydrate complexes. Appl. Microbiol. Biotechnol. 2019, 103, 4065–4075. [Google Scholar] [CrossRef]
- Shafiqur Rahman, M.; Qin, W. Glycosyltransferase Activity Assay Using Colorimetric Methods. Bact. Polysacch. Methods Protoc. 2019, 1954, 237–243. [Google Scholar]
- Yin, Y.; Chen, H.; Hahn, M.G.; Mohnen, D.; Xu, Y. Evolution and function of the plant cell wall synthesis-related glycosyltransferase family 8. Plant Physiol. 2010, 153, 1729–1746. [Google Scholar] [CrossRef]
- Linhardt, R.; Galliher, P.; Cooney, C. Polysaccharide lyases. Appl. Biochem. Biotechnol. 1987, 12, 135–176. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.; Latge, J.-P.; Munro, C.A. The fungal cell wall: Structure, biosynthesis, and function. Microbiol. Spectr. 2017, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Barman, A.; Gohain, D.; Bora, U.; Tamuli, R. Phospholipases play multiple cellular roles including growth, stress tolerance, sexual development, and virulence in fungi. Microbiol. Res. 2018, 209, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Beyond the Reference Genome; Nature Portfolio Heidelberger Platz 3: Berlin, Germany, 2023; Volume 616, pp. 618–620. [Google Scholar]
- Gournas, C.; Prévost, M.; Krammer, E.-M.; André, B. Function and regulation of fungal amino acid transporters: Insights from predicted structure. Yeast Membr. Transp. 2016, 892, 69–106. [Google Scholar]
- Sundaresan, A.K.; Gangwar, J.; Murugavel, A.; Malli Mohan, G.B.; Ramakrishnan, J. Complete genome sequence, phenotypic correlation and pangenome analysis of uropathogenic Klebsiella spp. AMB Express 2024, 14, 78. [Google Scholar] [CrossRef]
- Yi, R.; Mukaiyama, H.; Tachikawa, T.; Shimomura, N.; Aimi, T. A-mating-type gene expression can drive clamp formation in the bipolar mushroom Pholiota microspora (Pholiota nameko). Eukaryot. Cell 2010, 9, 1109–1119. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | P. nameko ZZ1 |
|---|---|
| Total Reads Count | 21,869,350 |
| Total Bases Count (bp) | 3,280,402,500 |
| Average Read Length (bp) | 150 |
| Q20 Bases Count (bp) | 3,219,066,634 |
| Q20 Bases Ratio | 98.13% |
| Q30 Bases Count (bp) | 3,129,325,413 |
| Q30 Bases Ratio | 95.39% |
| GC content | 45.25% |
| Genes | Strains | Number |
|---|---|---|
| Core genes | (90% ≤ strains ≤ 100%) | 2608 |
| Shell genes | (15% ≤ strains < 90%) | 9610 |
| Cloud genes | (1% ≤ strains < 15%) | 0 |
| Total genes | (0% ≤ strains ≤ 100%) | 12,218 |
| Latin Name | GeneBank | Pan | Core | Dispensable |
|---|---|---|---|---|
| Pholiota nameko | PQ839732 | 2743 | 2608 | 135 |
| Pholiota adiposa | GCA_009935795.1 | 3174 | 2608 | 564 |
| Pholiota microspora | GCA_003314615.1 | 2766 | 2608 | 158 |
| Pholiota molesta | GCA_014925825.1 | 3481 | 2608 | 873 |
| Pholiota conissans | GCA_015484465.1 | 3110 | 2608 | 502 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Liu, B.; Ouyang, X.; He, M.; Hui, H.; Tang, B.; Feng, L.; Ren, M.; Chen, G.; Liu, G.; et al. Whole-Genome Sequencing and Fine Map Analysis of Pholiota nameko. J. Fungi 2025, 11, 112. https://doi.org/10.3390/jof11020112
He Y, Liu B, Ouyang X, He M, Hui H, Tang B, Feng L, Ren M, Chen G, Liu G, et al. Whole-Genome Sequencing and Fine Map Analysis of Pholiota nameko. Journal of Fungi. 2025; 11(2):112. https://doi.org/10.3390/jof11020112
Chicago/Turabian StyleHe, Yan, Bo Liu, Xiaoqi Ouyang, Mianyu He, Hongyan Hui, Bimei Tang, Liaoliao Feng, Min Ren, Guoliang Chen, Guangping Liu, and et al. 2025. "Whole-Genome Sequencing and Fine Map Analysis of Pholiota nameko" Journal of Fungi 11, no. 2: 112. https://doi.org/10.3390/jof11020112
APA StyleHe, Y., Liu, B., Ouyang, X., He, M., Hui, H., Tang, B., Feng, L., Ren, M., Chen, G., Liu, G., & He, X. (2025). Whole-Genome Sequencing and Fine Map Analysis of Pholiota nameko. Journal of Fungi, 11(2), 112. https://doi.org/10.3390/jof11020112