
Induction of Extracellular Hydroxyl Radicals Production in the White-Rot Fungus Pleurotus eryngii for Dyes Degradation: An Advanced Bio-oxidation Process
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Dyes, Other Chemicals, and Enzymes
2.2. Organism and Culture Conditions
2.3. Determination of Fungal Biomass and Enzyme Activities
2.4. Quinone Redox Cycling Conditions for ·OH Production
2.5. Advanced Oxidation of Dyes
2.6. Analytical Techniques
2.7. Statistical Analyses
3. Results
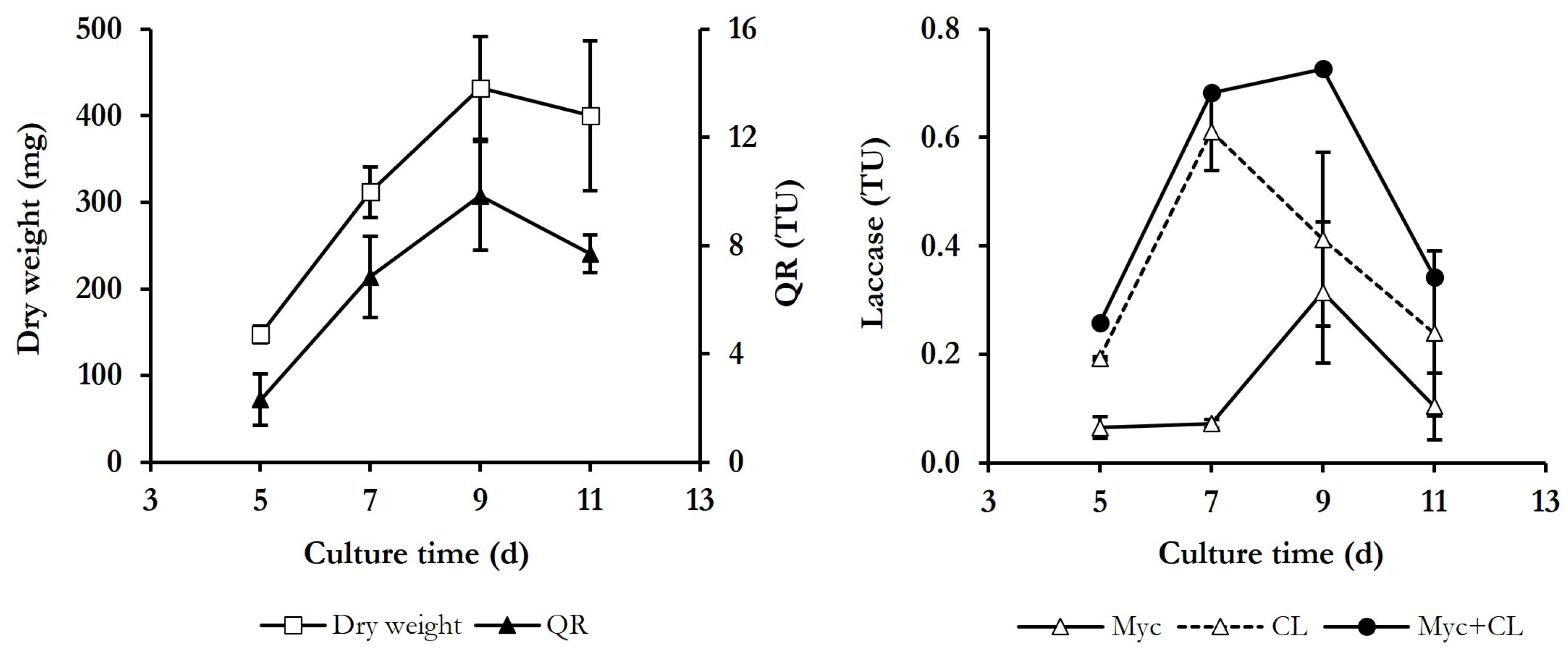
3.1. Determination of Culture Conditions for Both Dyes Decolourisation and Induction of ·OH Production
3.2. Decolourisation of Dyes by Fungal Culture and Laccase
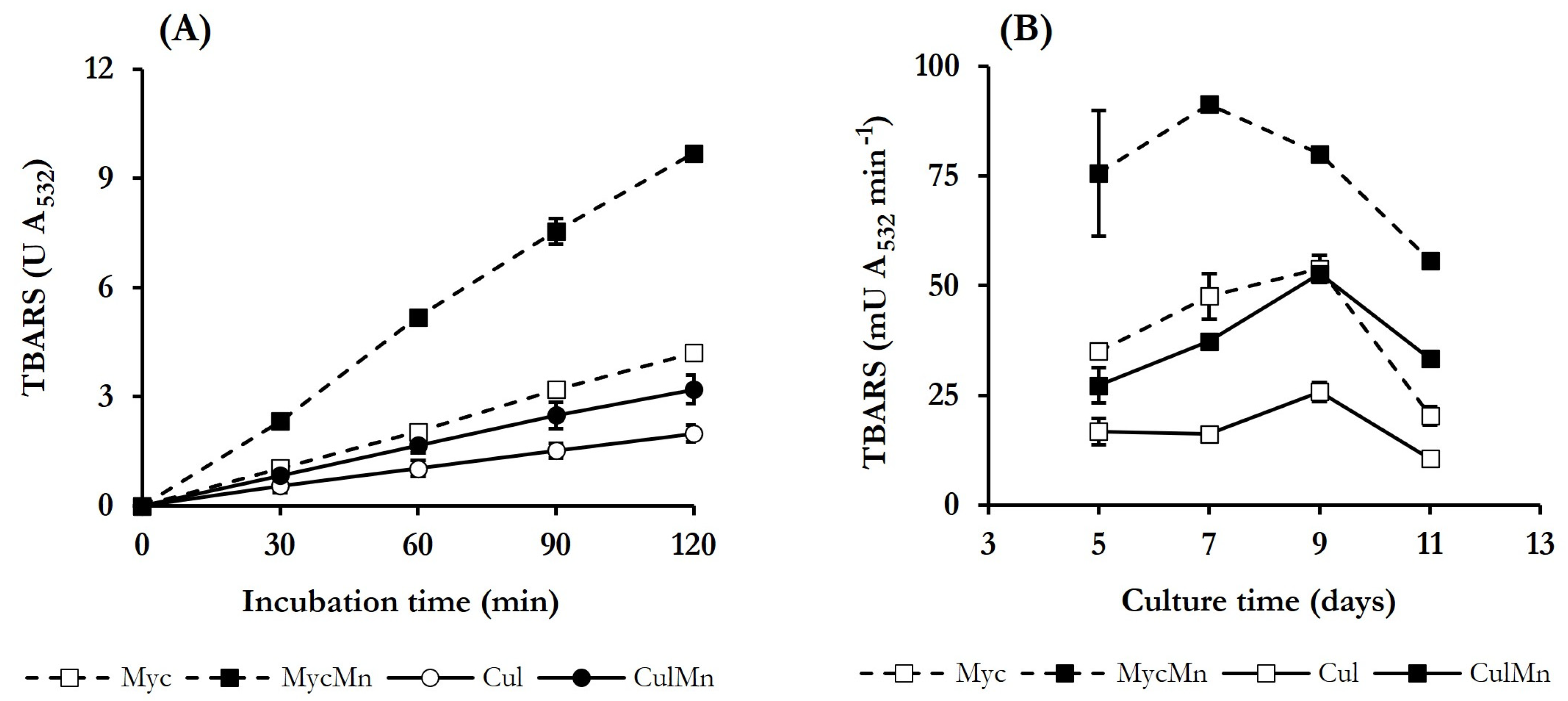
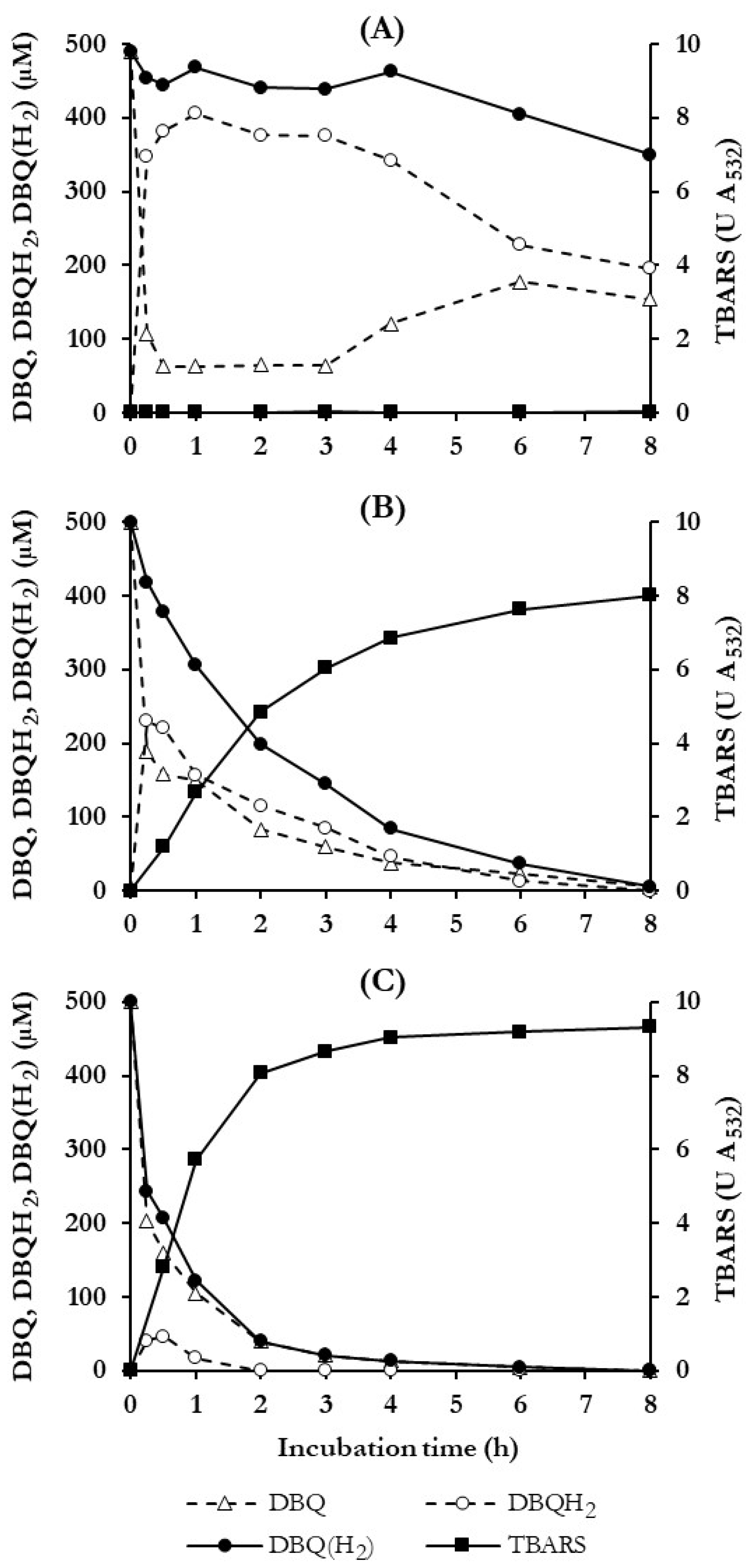
3.3. Selection of Quinone Redox Cycling Conditions for Induction of ·OH Production
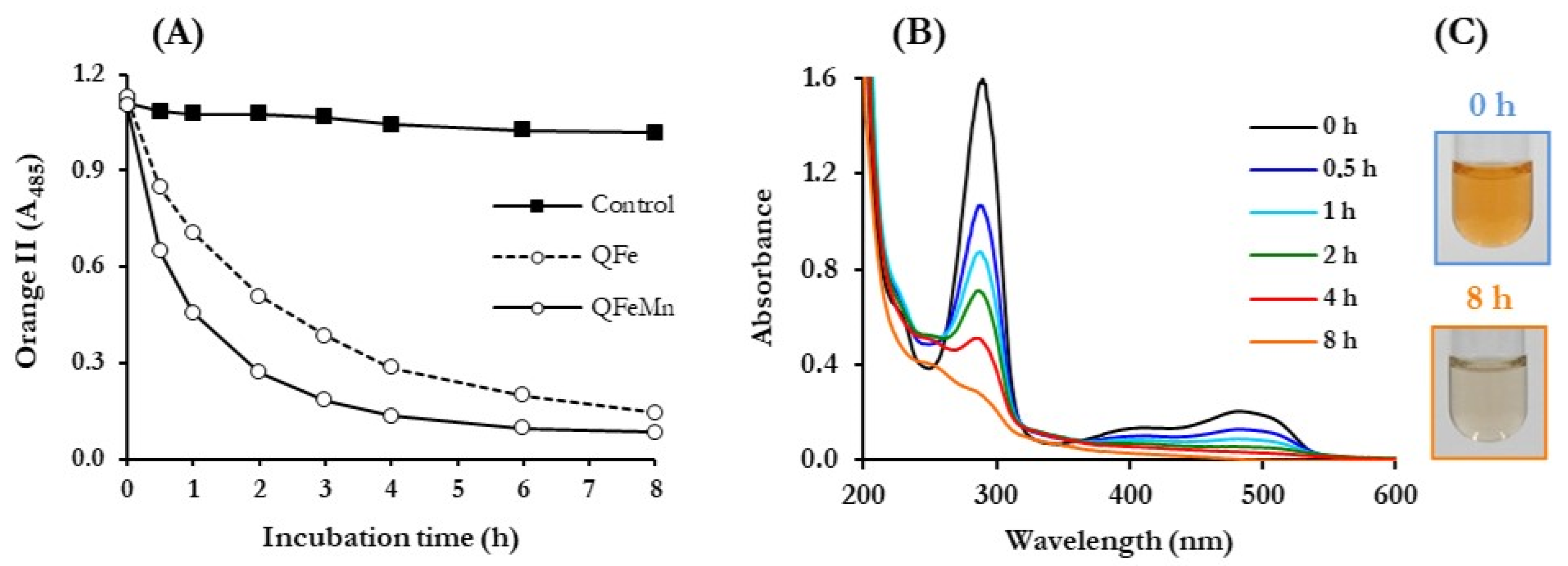
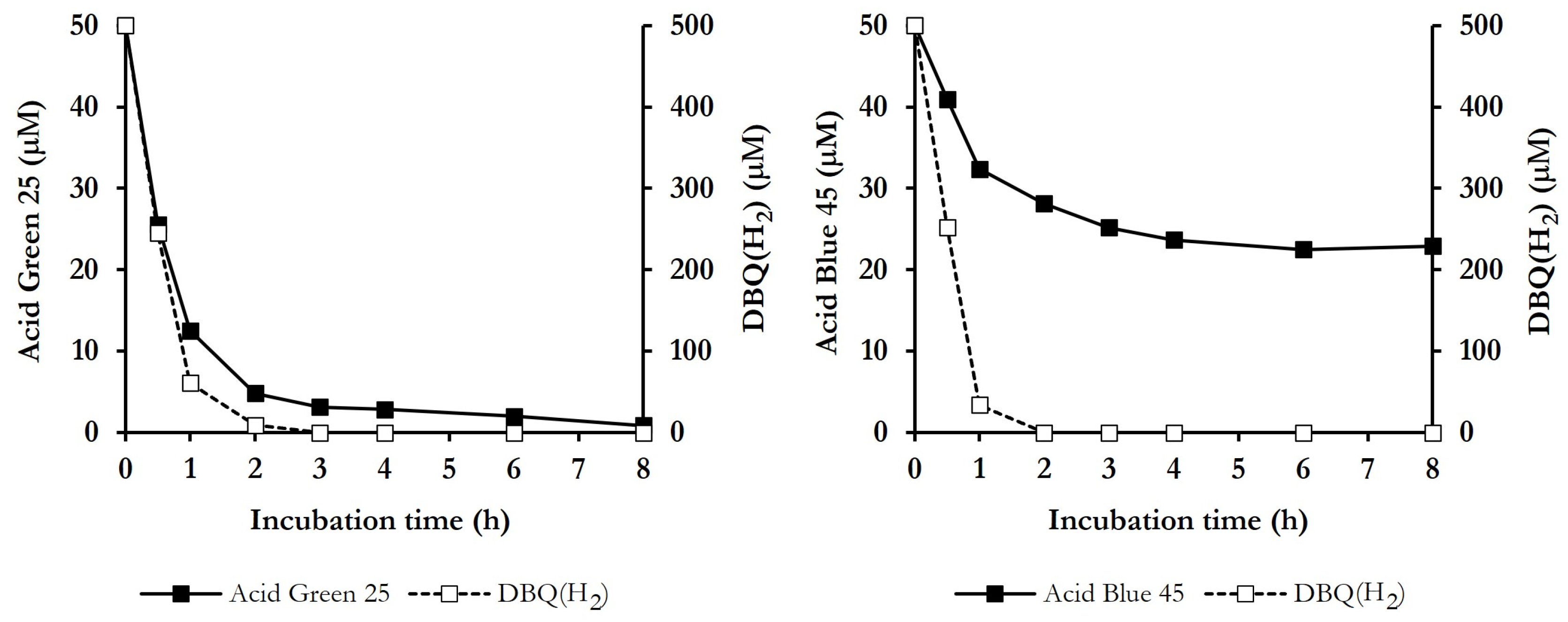
3.4. ABOP of Dyes, Mediated by P. eryngii
4. Discussion
4.1. From the Use of WRF and Their Enzymes for Pollutant Degradation to ABOP Mediated by These Fungi
4.2. ABOP of Dyes Mediated by P. eryngii
4.3. Perspectives and Future Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, D.S.; Yi, H.; Lai, C.; Liu, X.G.; Huo, X.Q.; An, Z.W.; Li, L.; Fu, Y.K.; Li, B.S.; Zhang, M.M.; et al. Critical review of advanced oxidation processes in organic wastewater treatment. Chemosphere 2021, 275, 130104. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, R.; Fan, F.F. A comprehensive insight into the application of white rot fungi and their lignocellulolytic enzymes in the removal of organic pollutants. Sci. Total Environ. 2021, 778, 146132. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, X.; Zhang, M.; Zhu, Y.H.; Zhuo, R. Removal of heavy-metal pollutants by white rot fungi: Mechanisms, achievements, and perspectives. J. Clean. Prod. 2022, 354, 131681. [Google Scholar] [CrossRef]
- Glaze, W.H.; Kang, J.W.; Chapin, D.H. The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation. Ozone-Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
- Parsons, S. Advanced Oxidation Processes for Water and Wastewater Treatment; IWA Publishing: London, UK, 2012. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment I: Oxidation technologies at ambient conditions. Adv. Environ. Res. 2004, 8, 501–551. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment II: Hybrid methods. Adv. Environ. Res. 2004, 8, 553–597. [Google Scholar] [CrossRef]
- Quiroz, M.A.; Bandala, E.R.; Martínez-Huitle, C.A. Advanced oxidation processes (AOPs) for removal of pesticides from aqueous media. In Pesticides—Formulations, Effects, Fate; Stoytcheva, M., Ed.; InTech Europe: Rijeka, Croacia, 2011; pp. 685–730. [Google Scholar] [CrossRef]
- Daifullah, A.H.A.; Mohamed, M.M. Degradation of benzene, toluene ethylbenzene and p-xylene (BTEX) in aqueous solutions using UV/H2O2 system. J. Chem. Technol. Biotechnol. 2004, 79, 468–474. [Google Scholar] [CrossRef]
- Ikehata, K.; El-Din, M.G. Degradation of recalcitrant surfactants in wasterwater by ozonation and advances oxidation processes: A review. Ozono-Sci. Eng. 2004, 26, 327–343. [Google Scholar] [CrossRef]
- Torrades, F.; García-Montaño, J.; García-Hortal, J.A.; Domènech, X.; Peral, J. Decolorization and mineralization of commercial reactive dyes under solar light assisted photo-Fenton conditions. Sol. Energy 2004, 77, 573–581. [Google Scholar] [CrossRef]
- Pera-Titus, M.; García-Molina, V.; Baños, M.A.; Giménez, J.; Esplugas, S. Degradation of chlorophenols by means of advanced oxidation processes: A general review. Appl. Catal. B Environ. 2004, 47, 219–256. [Google Scholar] [CrossRef]
- Heponiemi, A.; Lassi, U. Advanced oxidation processes in food industry wastewater treatment—A review. In Food Industrial Processes—Methods and Equipment; Valdez, B., Ed.; InTech Europe: Rijeta, Croacia, 2012; pp. 313–338. [Google Scholar] [CrossRef]
- Paraskeva, P.; Diamadopoulos, E. Technologies for olive mill wastewater (OMW) treatment: A review. J. Chem. Technol. Biotechnol. 2006, 81, 1475–1485. [Google Scholar] [CrossRef]
- Saien, J.; Nejati, H. Enhanced photocatalytic degradation of pollutants in petroleum refinery wastewater under mild conditions. J. Hazard. Mater. 2007, 148, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Pointing, S.B. Feasibility of bioremediation by white-rot fungi. Appl. Microbiol. Biotechnol. 2001, 57, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Tortella, G.R.; Diez, M.C.; Durán, N. Fungal diversity and use in decomposition of environmental pollutants. Crit. Rev. Microbiol. 2005, 31, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Cullen, D.; Kersten, P.J. Enzymology and molecular biology of lignin degradation. In The Mycota III. Biochemistry and Molecular Biology; Brambl, R., Marzluf, G.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 249–312. [Google Scholar] [CrossRef]
- Martínez, A.T.; Speranza, M.; Ruiz-Duenas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar] [PubMed]
- Schoemaker, H.E. On the chemistry of lignin degradation. Recl. Trav. Chim. Pays-Bas 1990, 109, 255–272. [Google Scholar] [CrossRef]
- Johannes, C.; Majcherczyk, A. Natural mediators in the oxidation of polycyclic aromatic hydrocarbons by laccase mediator systems. Appl. Environ. Microbiol. 2000, 66, 524–528. [Google Scholar] [CrossRef]
- Camarero, S.; Ibarra, D.; Martínez, M.J.; Martínez, A.T. Lignin-derived compounds as efficient laccase mediators for decolorization of different types of recalcitrant dyes. Appl. Environ. Microbiol. 2005, 71, 1775–1784. [Google Scholar] [CrossRef]
- Morozova, O.V.; Shumakovich, G.P.; Shleev, S.V.; Yaropolov, Y.I. Laccase-mediator systems and their applications: A review. Appl. Biochem. Microbiol. 2007, 43, 523–535. [Google Scholar] [CrossRef]
- Backa, S.; Gierer, J.; Reitberger, T.; Nilsson, T. Hydroxyl radical activity associated with the growth of white-rot fungi. Holzforschung 1993, 47, 181–187. [Google Scholar] [CrossRef]
- Joseleau, J.-P.; Gharibian, S.; Comtat, J.; Lefebvre, A.; Ruel, K. Indirect involvement of ligninolytic enzyme systems in cell wall degradation. FEMS Microbiol. Rev. 1994, 13, 255–264. [Google Scholar] [CrossRef][Green Version]
- Tanaka, H.; Itakura, S.; Enoki, A. Hydroxyl radical generation by an extracellular low-molecular-weight substance and phenol oxidase activity during wood degradation by the white-rot basidiomycete Phanerochaete chrysosporium. Holzforschung 1999, 53, 21–28. [Google Scholar] [CrossRef]
- Barr, D.P.; Shah, M.M.; Grover, T.A.; Aust, S.D. Production of hydroxyl radical by lignin peroxidase from Phanerochaete chrysosporium. Arch. Biochem. Biophys. 1992, 298, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Guillén, F.; Gómez-Toribio, V.; Martínez, M.J.; Martínez, A.T. Production of hydroxyl radical by the synergistic action of fungal laccase and aryl alcohol oxidase. Arch. Biochem. Biophys. 2000, 383, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.G.; Nicholls, P.; Wilson, M.T. Rotting by radicals—The role of cellobiose oxidoreductase? Biochem. Soc. Trans. 2003, 31, 1335–1336. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.L.; Ma, F.Y.; Chen, Q.; Xiao, Q.Y.; Zhang, X.Y.; Xie, S.X.; Yu, H.B. Lytic polysaccharide monooxygenases promote oxidative cleavage of lignin and lignin-carbohydrate complexes during fungal degradation of lignocellulose. Environ. Microbiol. 2021, 23, 4547–4560. [Google Scholar] [CrossRef] [PubMed]
- Guillén, F.; Muñoz, C.; Gómez-Toribio, V.; Martínez, A.T.; Martínez, M.J. Oxygen activation during the oxidation of methoxyhydroquinones by laccase from Pleurotus eryngii. Appl. Environ. Microbiol. 2000, 66, 170–175. [Google Scholar] [CrossRef]
- Peña, A.; Babiker, R.; Chaduli, D.; Lipzen, A.; Wang, M.; Chovatia, M.; Rencoret, J.; Marques, G.; Sánchez-Ruiz, M.I.; Kijpornyongpan, T.; et al. A multiomic approach to understand how Pleurotus eryngii transforms non-woody lignocellulosic material. J. Fungi 2021, 7, 426. [Google Scholar] [CrossRef]
- Gómez-Toribio, V.; García-Martín, A.B.; Martínez, M.J.; Martínez, A.T.; Guillén, F. Induction of extracellular hydroxyl radical production by white-rot fungi through quinone redox cycling. Appl. Environ. Microbiol. 2009, 75, 3944–3953. [Google Scholar] [CrossRef]
- Gómez-Toribio, V.; García-Martín, A.B.; Martínez, M.J.; Martínez, A.T.; Guillén, F. Enhancing the production of hydroxyl radicals by Pleurotus eryngii via quinone redox cycling for pollutant removal. Appl. Environ. Microbiol. 2009, 75, 3954–3962. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Oliveros, E.; MacKay, A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit. Rev. Environ. Sci. Technol. 2006, 36, 1–84. [Google Scholar] [CrossRef]
- Chen, S.X.; Zhu, M.D.; Guo, X.Y.; Yang, B.T.; Zhuo, R. Coupling of Fenton reaction and white rot fungi for the degradation of organic pollutants. Ecotoxicol. Environ. Saf. 2023, 254, 114697. [Google Scholar] [CrossRef] [PubMed]
- Baker, W. Derivatives of pentahydroxybenzene, and a synthesis of pedicellin. J. Chem. Soc. 1941, 1941, 662–670. [Google Scholar] [CrossRef]
- Muñoz, C.; Guillén, F.; Martínez, A.T.; Martínez, M.J. Laccase isoenzymes of Pleurotus eryngii: Characterization, catalytic properties and participation in activation of molecular oxygen and Mn2+ oxidation. Appl. Environ. Microbiol. 1997, 63, 2166–2174. [Google Scholar] [CrossRef]
- Marco-Urrea, E.; Aranda, E.; Caminal, G.; Guillén, F. Induction of hydroxyl radical production in Trametes versicolor to degrade recalcitrant chlorinated hydrocarbons. Bioresour. Technol. 2009, 100, 5757–5762. [Google Scholar] [CrossRef] [PubMed]
- Aranda, E.; Marco-Urrea, E.; Caminal, G.; Arias, M.E.; García-Romera, I.; Guillén, F. Advanced oxidation of benzene, toluene, ethylbenzene and xylene isomers (BTEX) by Trametes versicolor. J. Hazard. Mater. 2010, 181, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Formation of a thiobarbituric-acid-reactive substance from deoxyribose in the presence of iron salts. FEBS Lett. 1981, 128, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Bezalel, L.; Hadar, Y.; Cerniglia, C.E. Enzymatic mechanisms involved in phenanthrene degradation by the white rot fungus Pleurotus ostreatus. Appl. Environ. Microbiol. 1997, 63, 2495–2501. [Google Scholar] [CrossRef]
- Rieble, S.; Joshi, D.K.; Gold, M.H. Purification and characterization of a 1,2,4-trihydroxybenzene 1,2-dioxygenase from the basidiomycete Phanerochaete chrysosporium. J. Bacteriol. 1994, 176, 4838–4844. [Google Scholar] [CrossRef]
- Bumpus, J.A.; Tien, M.; Wright, D.; Aust, S.D. Oxidation of persistent environmental pollutants by a white rot fungus. Science 1985, 228, 1434–1436. [Google Scholar] [CrossRef]
- Lebkowska, M.; Zaleska-Radziwill, M. Application of white-rot fungi for biodegradation of refractory organic compounds-a review. Desalin. Water Treat. 2014, 52, 3708–3713. [Google Scholar] [CrossRef]
- Cabana, H.; Jones, J.P.; Agathos, S.N. Elimination of endocrine disrupting chemicals using white rot fungi and their lignin modifying enzymes: A review. Eng. Life Sci. 2007, 7, 429–456. [Google Scholar] [CrossRef]
- Cvancarova, M.; Kresinova, Z.; Filipova, A.; Covino, S.; Cajthaml, T. Biodegradation of PCBs by ligninolytic fungi and characterization of the degradation products. Chemosphere 2012, 88, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.B.; Hai, F.I.; Singh, L.; Price, W.E.; Nghiem, L.D. Degradation of pharmaceuticals and personal care products by white-rot fungi-a critical review. Curr. Pollut. Rep. 2017, 3, 88–103. [Google Scholar] [CrossRef]
- Herath, I.S.; Udayanga, D.; Jayasanka, D.J.; Hewawasam, C. Textile dye decolorization by white rot fungi—A review. Bioresour. Technol. Rep. 2024, 25, 101687. [Google Scholar] [CrossRef]
- Cameron, M.D.; Timofeevski, S.; Aust, S.D. Enzymology of Phanerochaete chrysosporium with respect to the degradation of recalcitrant compounds and xenobiotics. Appl. Microbiol. Biotechnol. 2000, 54, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Stajic, M.; Vukojevic, J.; Duletic-Lausevic, S. Biology of Pleurotus eryngii and role in biotechnological processes: A review. Crit. Rev. Biotechnol. 2009, 29, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Bilal, M.; Iqbal, H.M.N.; Raj, A. Lignin peroxidase in focus for catalytic elimination of contaminants? A critical review on recent progress and perspectives. Int. J. Biol. Macromol. 2021, 177, 58–82. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Kaur, N.; Selvaraj, M.; Ghramh, H.A.; Al-Shehri, B.M.; Singh, G.; Arya, S.K.; Bhatt, K.; Ghotekar, S.; Mani, R.; et al. Laccase-assisted degradation of emerging recalcitrant compounds—A review. Bioresour. Technol. 2022, 364, 128031. [Google Scholar] [CrossRef]
- Saikia, S.; Yadav, M.; Hoque, R.A.; Yadav, H.S. Bioremediation mediated by manganese peroxidase—An overview. Biocatal. Biotransform. 2023, 41, 161–173. [Google Scholar] [CrossRef]
- Schoemaker, H.E.; Meijer, E.M.; Leisola, M.S.A.; Haemmerli, S.D.; Waldner, R.; Sanglard, D.; Schmidt, H.W.H. Oxidation and reduction in lignin biodegradation. In Plant Cell Wall Polymers: Biogenesis and Biodegradation (ACS Symp. Ser. 399); Lewis, N.G., Paice, M.G., Eds.; American Chemical Society: Washington, DC, USA, 1989; pp. 454–471. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, C.A. Degradation of benzene, toluene, ethylbenzene, and xylenes (BTEX) by the lignin-degrading basidiomycete Phanerochaete chrysosporium. Appl. Environ. Microbiol. 1993, 59, 756–762. [Google Scholar] [CrossRef]
- Marco-Urrea, E.; Radjenovic, J.; Caminal, G.; Petrovic, M.; Vicent, T.; Barceló, D. Oxidation of atenolol, propranolol, carbamazepine and clofibric acid by a biological Fenton-like system mediated by the white-rot fungus Trametes versicolor. Water Res. 2010, 44, 521–532. [Google Scholar] [CrossRef] [PubMed]
- del Alamo, A.C.; Pariente, M.I.; Molina, R.; Martinez, F. Advanced bio-oxidation of fungal mixed cultures immobilized on rotating biological contactors for the removal of pharmaceutical micropollutants in a real hospital wastewater. J. Hazard. Mater. 2022, 425, 128002. [Google Scholar] [CrossRef]
- Vasiliadou, I.A.; Molina, R.; Pariente, M.I.; Christoforidis, K.C.; Martinez, F.; Melero, J.A. Understanding the role of mediators in the efficiency of advanced oxidation processes using white-rot fungi. Chem. Eng. J. 2019, 359, 1427–1435. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Caramelo, L.; Prieto, A.; Martínez, M.J.; Martínez, A.T. Anisaldehyde production and aryl-alcohol oxidase and dehydrogenase activities in ligninolytic fungi from the genus Pleurotus. Appl. Environ. Microbiol. 1994, 60, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Heinfling, A.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Bergbauer, M.; Szewzyk, U.; Martínez, A.T. A study on reducing substrates of manganese-oxidizing peroxidases from Pleurotus eryngii and Bjerkandera adusta. FEBS Lett. 1998, 428, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Nidheesh, P.V.; Gandhimathi, R.; Ramesh, S.T. Degradation of dyes from aqueous solution by Fenton processes: A review. Environ. Sci. Pollut. Res. 2013, 20, 2099–2132. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Decolourisation (%) | |||
|---|---|---|---|
| Structure | Dye | Culture | Laccase |
| Anthraquinone | Acid Black 48 | Adsorption | 17 ± 1 |
| Acid Blue 45 | 14 ± 1 | 7 ± 0 | |
| Acid Green 25 | 57 ± 3 | 86 ± 2 | |
| Reactive Blue 19 | 22 ± 2 | 24 ± 1 | |
| Azo | Acid Red 88 | Adsorption | 7 ± 0 |
| Acid Yellow 17 | 0 ± 0 | 0 ± 0 | |
| Chromotrope 2R | 29 ± 2 | 6 ± 1 | |
| Crocein Orange G | 19 ± 2 | 2 ± 0 | |
| Methyl Orange | 22 ± 3 | 4 ± 0 | |
| New Coccine | 41 ± 3 | 0 ± 0 | |
| Orange II | 38 ± 4 | 3 ± 1 | |
| Tartrazine | 0 ± 0 | 0 ± 0 | |
| Tropaeolin O | Adsorption | 2 ± 0 | |
| Diazo | Acid Black 24 | Adsorption | 53 ± 2 |
| Acid Blue 113 | Adsorption | 73 ± 2 | |
| Acid Orange 63 | Adsorption | 0 ± 0 | |
| Ponceau SS | 12 ± 1 | 0 ± 0 | |
| Reactive Black 5 | 1 ± 0 | 15 ± 1 | |
| Heterocycle | Azure B | Adsorption | 39 ± 2 |
| Neutral Red | Adsorption | 0 ± 0 | |
| Indigo | Indigo Carmine | 76 ± 4 | 2 ± 0 |
| Phthalocyanine | Iron(III)phthalocyanine | 44 ± 3 | 67 ± 2 |
| Triarylmethane | Brilliant Green | 85 ± 3 | 61 ± 2 |
| Bromophenol Blue | 3 ± 1 | 27 ± 1 | |
| Cresol Red | Adsorption | 40 ± 2 | |
| Crystal Violet | Adsorption | 13 ± 1 | |
| Methyl Blue | 17 ± 2 | 0 ± 0 | |
| Decolourisation (%) | Degradation (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Structure | Dye | QFe | QFeMn | QFeMn | ||||
| 1 h | 3 h | 8 h | 1 h | 3 h | 8 h | 8 h | ||
| Anthraquinone | Acid Black 48 1 | Mycelial adsorption | ||||||
| Acid Blue 45 | 16 ± 6 | 20 ± 6 | 22 ± 2 | 17 ± 4 | 23 ± 2 | 25 ± 5 | 54 (8 h) | |
| Acid Green 25 | 38 ± 3 | 61 ± 3 | 68 ± 6 | 48 ± 5 | 67 ± 3 | 65 ± 1 | 98 (8 h) | |
| Reactive Blue 19 | 18 ± 2 | 30 ± 3 | 50 ± 7 | 28 ± 6 | 42 ± 7 | 52 ± 4 | 93 (8 h) | |
| Azo | Acid Red 88 1 | 46 ± 5 | 79 ± 3 | 83 ± 6 | 45 ± 2 | 79 ± 4 | 89 ± 4 | 100 (4 h) |
| Acid Yellow 17 | 30 ± 2 | 68 ± 1 | 90 ± 1 | 50 ± 2 | 87 ± 1 | 91 ± 0 | 99 (8 h) | |
| Chromotrope 2R | 29 ± 4 | 67 ± 2 | 87 ± 2 | 50 ± 5 | 81 ± 1 | 91 ± 1 | 99 (8 h) | |
| Crocein Orange G | 38 ± 2 | 67 ± 1 | 86 ± 1 | 60 ± 6 | 82 ± 3 | 90 ± 1 | 100 (4 h) | |
| Methyl Orange | 22 ± 4 | 56 ± 5 | 81 ± 0 | 48 ± 3 | 82 ± 1 | 87 ± 1 | 100 (3 h) | |
| New Coccine | 37 ± 4 | 78 ± 3 | 94 ± 0 | 68 ± 6 | 91 ± 1 | 95 ± 0 | 49 (3 h) | |
| Orange II | 37 ± 2 | 66 ± 5 | 87 ± 3 | 59 ± 4 | 83 ± 7 | 92 ± 1 | 99 (8 h) | |
| Tartrazine | 30 ± 4 | 56 ± 3 | 82 ± 2 | 45 ± 3 | 78 ± 4 | 90 ± 2 | 100 (3 h) | |
| Tropaeolin O | 28 ± 2 | 55 ± 3 | 78 ± 1 | 34 ± 1 | 68 ± 3 | 85 ± 2 | 100 (3 h) | |
| Diazo | Acid Black 24 1 | 9 ± 2 | 42 ± 2 | 72 ± 2 | 15 ± 3 | 54 ± 3 | 78 ± 2 | 95 (8 h) |
| Acid Blue 113 1 | Mycelial adsorption | |||||||
| Acid Orange 63 1 | 39 ± 5 | 65 ± 3 | 84 ± 4 | 50 ± 3 | 74 ± 2 | 89 ± 0 | 89 (8 h) | |
| Ponceau SS | 37 ± 1 | 68 ± 3 | 84 ± 2 | 56 ± 3 | 86 ± 3 | 89 ± 2 | 100 (4 h) | |
| Reactive Black 5 | 34 ± 4 | 66 ± 5 | 86 ± 1 | 53 ± 3 | 83 ± 4 | 90 ± 1 | 100 (2 h) | |
| Heterocycle | Azure B 1 | Mycelial adsorption | ||||||
| Neutral Red 1 | Mycelial adsorption | |||||||
| Indigo | Indigo Carmine | 74 ± 1 | 96 ± 0 | 97 ± 0 | 100 | 100 ± 0 | 100 ± 0 | 100 (1 h) |
| Phthalocyanine | Iron(III)phthaloc. | 64 ± 4 | 75 ± 1 | 82 ± 1 | 72 ± 2 | 82 ± 5 | 83 ± 4 | nd (8 h) |
| Triarylmehane | Brilliant Green | 54 ± 2 | 78 ± 4 | 94 ± 3 | 59 ± 5 | 80 ± 4 | 93 ± 8 | 100 (6 h) |
| Bromophenol Blue | 34 ± 2 | 57 ± 1 | 77 ± 3 | 52 ± 1 | 73 ± 1 | 77 ± 3 | 91 (8 h) | |
| Cresol Red 1 | 54 ± 1 | 87 ± 3 | 93 ± 1 | 74 ± 6 | 91 ± 3 | 93 ± 1 | 100 (8 h) | |
| Crystal Violet 1 | Mycelial adsorption | |||||||
| Methyl Blue | 35 ± 2 | 54 ± 1 | 71 ± 3 | 40 ± 1 | 62 ± 4 | 74 ± 3 | 89 (8 h) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Martín, A.B.; Rodríguez, J.; Molina-Guijarro, J.M.; Fajardo, C.; Domínguez, G.; Hernández, M.; Guillén, F. Induction of Extracellular Hydroxyl Radicals Production in the White-Rot Fungus Pleurotus eryngii for Dyes Degradation: An Advanced Bio-oxidation Process. J. Fungi 2024, 10, 52. https://doi.org/10.3390/jof10010052
García-Martín AB, Rodríguez J, Molina-Guijarro JM, Fajardo C, Domínguez G, Hernández M, Guillén F. Induction of Extracellular Hydroxyl Radicals Production in the White-Rot Fungus Pleurotus eryngii for Dyes Degradation: An Advanced Bio-oxidation Process. Journal of Fungi. 2024; 10(1):52. https://doi.org/10.3390/jof10010052
Chicago/Turabian StyleGarcía-Martín, Ana Belén, Juana Rodríguez, José Manuel Molina-Guijarro, Carmen Fajardo, Gabriela Domínguez, Manuel Hernández, and Francisco Guillén. 2024. "Induction of Extracellular Hydroxyl Radicals Production in the White-Rot Fungus Pleurotus eryngii for Dyes Degradation: An Advanced Bio-oxidation Process" Journal of Fungi 10, no. 1: 52. https://doi.org/10.3390/jof10010052
APA StyleGarcía-Martín, A. B., Rodríguez, J., Molina-Guijarro, J. M., Fajardo, C., Domínguez, G., Hernández, M., & Guillén, F. (2024). Induction of Extracellular Hydroxyl Radicals Production in the White-Rot Fungus Pleurotus eryngii for Dyes Degradation: An Advanced Bio-oxidation Process. Journal of Fungi, 10(1), 52. https://doi.org/10.3390/jof10010052