
It Is Not Carved in Stone—The Need for a Genetic Reevaluation of Variants in Pediatric Cardiomyopathies

 , , and
, , and
Abstract
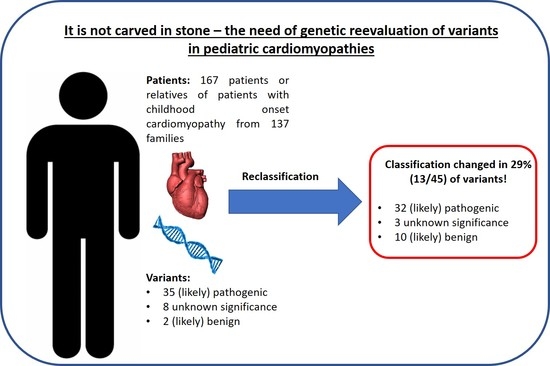
:
1. Introduction
2. Materials and Methods
2.1. Clinical Study
2.2. Variant Interpretation
2.3. Statistics
3. Results
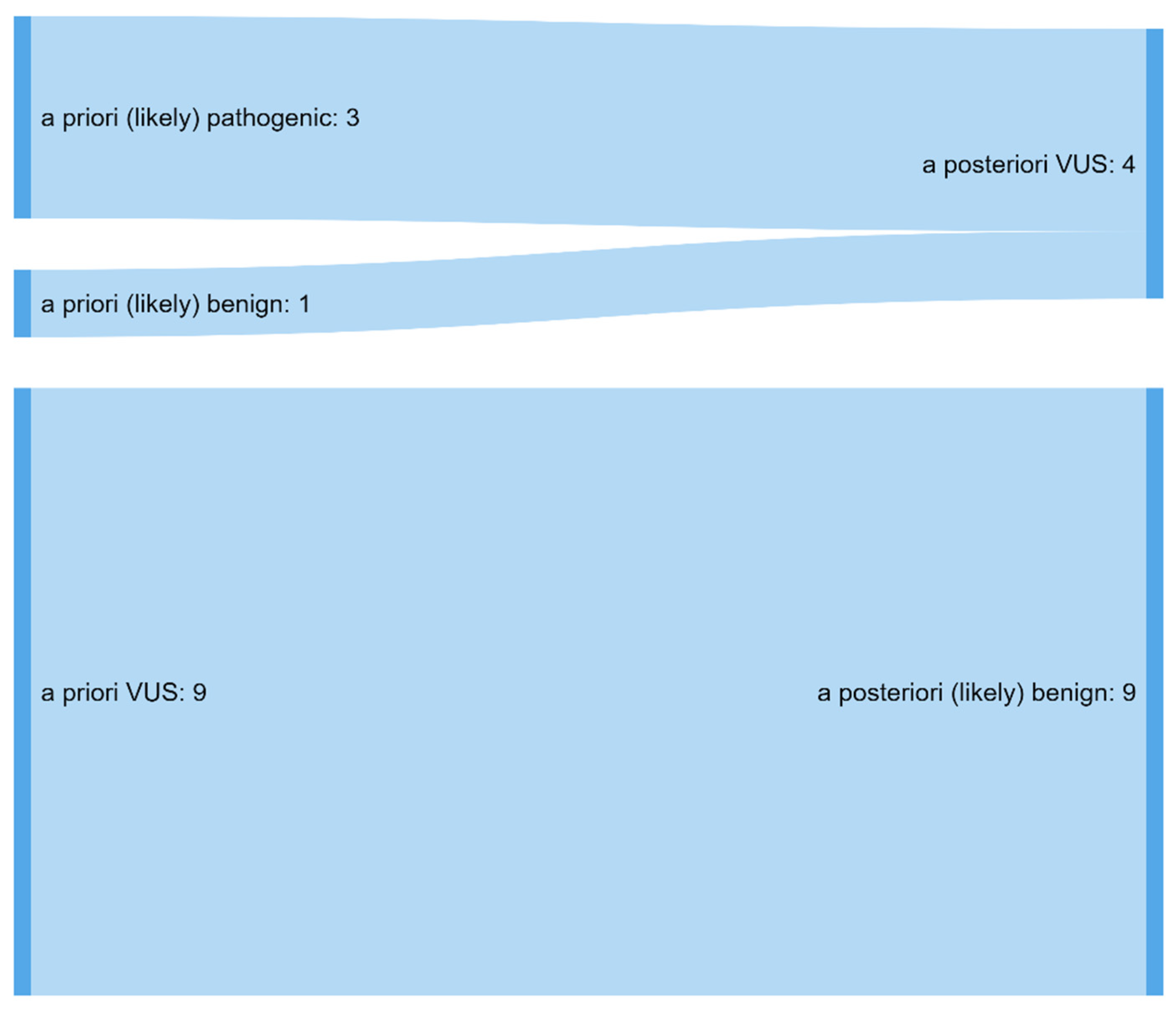
3.1. A Total of 29% of Variants Were Significantly Reclassified
3.2. The Number of Detected Variants in One Patient Correlates with the Chance of Reclassification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilescu, C.; Ojala, T.H.; Brilhante, V.; Ojanen, S.; Hinterding, H.M.; Palin, E.; Alastalo, T.P.; Koskenvuo, J.; Hiippala, A.; Jokinen, E.; et al. Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J. Am. Coll. Cardiol. 2018, 72, 2324–2338. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinrinade, O.; Ollila, L.; Vattulainen, S.; Tallila, J.; Gentile, M.; Salmenpera, P.; Koillinen, H.; Kaartinen, M.; Nieminen, M.S.; Myllykangas, S.; et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur. Heart J. 2015, 36, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.S.; Burkard, T.; Moscu-Gregor, A.; Gebauer, R.; Hessling, G.; Wolf, C.M. Reclassification of genetic variants in children with long QT syndrome. Mol. Genet. Genom. Med. 2020, 8, e1300. [Google Scholar] [CrossRef]
- Das, K.J.; Ingles, J.; Bagnall, R.D.; Semsarian, C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: Importance of periodic reassessment. Genet. Med. 2014, 16, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Vallverdu-Prats, M.; Alcalde, M.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Fernandez-Falgueras, A.; Coll, M.; Perez-Serra, A.; Puigmule, M.; Iglesias, A.; et al. Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. J. Pers. Med. 2021, 11, 162. [Google Scholar] [CrossRef]
- Quiat, D.; Witkowski, L.; Zouk, H.; Daly, K.P.; Roberts, A.E. Retrospective Analysis of Clinical Genetic Testing in Pediatric Primary Dilated Cardiomyopathy: Testing Outcomes and the Effects of Variant Reclassification. J. Am. Heart Assoc. 2020, 9, e016195. [Google Scholar] [CrossRef]
- Towbin, J.A. Pediatric Primary Dilated Cardiomyopathy Gene Testing and Variant Reclassification: Does It Matter? J. Am. Heart Assoc. 2020, 9, e016910. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working, G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Mazzarotto, F.; Whiffin, N.; Buchan, R.; Midwinter, W.; Wilk, A.; Li, N.; Felkin, L.; Ingold, N.; Govind, R.; et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Med. 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Tugrul, O.F.; Lai, A.; Amr, A.; Haas, J.; Proctor, T.; Ehlermann, P.; Jensen, K.; Katus, H.A.; et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: A meta-analysis on 7675 individuals. Clin. Res. Cardiol. 2018, 107, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Pirruccello, J.P.; Bick, A.; Wang, M.; Chaffin, M.; Friedman, S.; Yao, J.; Guo, X.; Venkatesh, B.A.; Taylor, K.D.; Post, W.S.; et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 2020, 11, 2254. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef]
- Manrai, A.K.; Funke, B.H.; Rehm, H.L.; Olesen, M.S.; Maron, B.A.; Szolovits, P.; Margulies, D.M.; Loscalzo, J.; Kohane, I.S. Genetic Misdiagnoses and the Potential for Health Disparities. N. Engl. J. Med. 2016, 375, 655–665. [Google Scholar] [CrossRef]
- Morales, A.; Kinnamon, D.D.; Jordan, E.; Platt, J.; Vatta, M.; Dorschner, M.O.; Starkey, C.A.; Mead, J.O.; Ai, T.; Burke, W.; et al. Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ. Genom. Precis. Med. 2020, 13, e002480. [Google Scholar] [CrossRef]
- Deo, R.C. Alternative Splicing, Internal Promoter, Nonsense-Mediated Decay, or All Three: Explaining the Distribution of Truncation Variants in Titin. Circ. Cardiovasc. Genet. 2016, 9, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, J.; Norambuena, P.; Tomasov, P.; Jindrova, D.; Sediva, H.; Macek, M., Jr.; Veselka, J. The utility of the Mayo Score for predicting the yield of genetic testing in patients with hypertrophic cardiomyopathy. Arch. Med. Sci. 2019, 15, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.K.; Bartels, K.; Hathaway, J.; Burns, C.; Yeates, L.; Semsarian, C.; Krahn, A.D.; Virani, A.; Ingles, J. Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur. J. Hum. Genet. 2019, 27, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee Members; Silka, M.J.; Shah, M.J.; Silva, J.N.A.; Balaji, S.; Beach, C.M.; Benjamin, M.N.; Berul, C.I.; Cannon, B.; Cecchin, F.; et al. 2021 PACES Expert Consensus Statement on the Indications and Management of Cardiovascular Implantable Electronic Devices in Pediatric Patients: Executive Summary. Heart Rhythm 2021, 18, 1925–1950. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 2015, 36, 2757–2759. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart J. 2021, 42, 563–645. [Google Scholar] [CrossRef]
- Bosman, L.P.; Te Riele, A. Arrhythmogenic right ventricular cardiomyopathy: A focused update on diagnosis and risk stratification. Heart 2022, 108, 90–97. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| N = 167 | HCM n = 75 | DCM n = 12 | ACM n = 3 | LVNC n = 12 | RCM n = 2 | Noonan Syndrome/NSML n = 26 | Morbus Fabry n = 1 | No Clinical Phenotype n = 36 |
|---|---|---|---|---|---|---|---|---|
| Male n = 101 | 48 | 7 | 2 | 8 | 1 | 16 | 0 | 19 |
| Positive family history n = 102 | 44 | 7 | 2 | 6 | 2 | 4 | 1 | 36 |
| Patients without genetic testing n = 41 | 15 | 4 | 1 | 3 | 0 | 3 | 0 | 15 |
| Patients with positive genetic findings n = 71 | 30 | 6 | 2 | 7 | 0 | 15 | 0 | 11 |
| Initial classification of variants N = 45 * | ||||||||
| Likely benign (class II) n = 2 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| VUS (class III) n = 8 | 2 | 3 | 0 | 2 | 0 | 0 | 0 | 1 |
| Likely pathogenic (class IV) n = 14 | 9 | 2 | 0 | 1 | 0 | 1 | 0 | 1 |
| Pathogenic (class V) n = 21 | 9 | 0 | 1 | 1 | 0 | 8 | 0 | 2 |
| Variable | All Patients (n = 71) | No Reclassification (n = 59) | Reclassification (n = 7) † | p-Value |
|---|---|---|---|---|
| Age at first diagnosis (years) * | 4.7 [0–50.3] | 5.5 [0–50.3] | 0 [0–12] | 0.219 |
| Length between first diagnosis and last clinical follow-up (years) * | 7.7 [0–33.5] | 6.1 [80–33.5] | 15.9 [7.4–26] | 0.047 |
| NYHA/Ross * | 1 [1–2.5] | 1 [1–2.5] | 1.5 [1–1.5] | 0.596 |
| Ejection fraction (%) * | 70 [25–94] | 70 [25–94] | 69 [61–81] | 0.863 |
| Number of cardiac medications * | 0 [0–5] | 0 [0–5] | 0.5 [0–2] | 0.805 |
| Death | No deaths | |||
| Hospital stay **,† | 26/58 (44.8%) | 24/52 (46.2%) | 2/6 (33.3%) | 0.681 |
| ICD implantation **,† | ||||
| Primary prophylactic | 7/68 (10.3%) | 6/61 (9.8%) | 1/7 (14.3%) | 0.550 |
| Secondary prophylactic | None | |||
| Appropriate ICD discharge **,† | 5/7 (7.1%) | 5/6 (83.3%) | 1/1 (100%) | 0.286 |
| Cardiac surgery **,† | 13/68 (19.1%) | 11/61 (18%) | 2/7 (28.6%) | 0.611 |
| Patient ID | Phenotype | Variant | Original Classification | Reclassification | Applied ACMG Criteria [6] | Year of Initial Report | Further Variants |
|---|---|---|---|---|---|---|---|
| 31 | HCM | TPM1 (NM_001018005.1): c.287A > G, p.(Glu96Gly) | LP | VUS | PM1, PP3, PP5 | 2014 | - |
| 49 | LVNC | TTN (NM_001267550.2): c.97612C > T, p.(Arg32538Cys) | VUS | LB | BS1, BP6 | 2019 | VUS in TTN, B variant in LMNA |
| 49 | LVNC | LMNA (NM_170707.3): c.1930C > T, p.(Arg644Cys) | VUS | B | PS3, BS1, BS4, BP6 | 2019 | VUS and LB variant in TTN |
| 60 | HCM | MYBPC3 (NM_000256.3): c.1468G > A, p.(Gly490Arg) ** | LP | VUS | PP3, PP5, BS1 | unknown | - |
| 61 | HCM | MYBPC3 (NM_000256.3): c.405A > G, p.(Lys135 = ) | VUS | LB | BS3, BP6, PM2 | 2018 | - |
| 65 | DCM | TTN (NM_001267550.2): c.94851T > A, p.(Asp31617Glu) | VUS | LB | BS1, BP4, BP6 | 2015 | LB and B variants in TTN |
| 65 | DCM | TTN (NM_001267550.2): c.14870C > G, p.(Thr4957Ser) | VUS | LB | BS1, BP6 | 2015 | LB and B variants in TTN |
| 65 | DCM | DSG2 (NM_001943.3): c.1174G > A, p.(Val392Ile) | VUS | B | BS1, BS3, BP4, BP6 | 2015 | Two LB variants in TTN |
| 121 | HCM | DSP (NM_004415.4): c.7655T > C, p.(Leu2552Pro) | LB | VUS | PM2, PP3 | 2018 | - |
| 124 | HCM | MYBPC3 (NM_000256.3): c.2992C > G, p.(Gln998Glu) | VUS | LB | PP2, BS1, BP6 | unknown | - |
| 125, 145, 172 * | DCM | MYOZ2 (NM_016599.5): c.713G > T, p.(Gly238Val) | LP | VUS | PM2, PP3 | 2015 | P variant in DSP |
| 146 | HCM | MYBPC3 (NM_000256.3): c.1468G > A, p.(Gly490Arg) ** | LP | VUS | PP3, PP5, BS1 | 2016 | - |
| 168 | HCM | TTN (NM_001267550.2): c.19738C > T, p.(Pro6580Ser) | VUS | LB | BS1, BP6 | 2017 | VUS in MYH6, LB variant in TTN |
| 168 | HCM | TTN (NM_001267550.2): c.18961A > G, p.(Ile6321Val) | VUS | LB | BS1, BP4, BP6 | 2017 | VUS in MYH6, LB variant in TTN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westphal, D.S.; Pollmann, K.; Marschall, C.; Wacker-Gussmann, A.; Oberhoffer-Fritz, R.; Laugwitz, K.-L.; Ewert, P.; Wolf, C.M. It Is Not Carved in Stone—The Need for a Genetic Reevaluation of Variants in Pediatric Cardiomyopathies. J. Cardiovasc. Dev. Dis. 2022, 9, 41. https://doi.org/10.3390/jcdd9020041
Westphal DS, Pollmann K, Marschall C, Wacker-Gussmann A, Oberhoffer-Fritz R, Laugwitz K-L, Ewert P, Wolf CM. It Is Not Carved in Stone—The Need for a Genetic Reevaluation of Variants in Pediatric Cardiomyopathies. Journal of Cardiovascular Development and Disease. 2022; 9(2):41. https://doi.org/10.3390/jcdd9020041
Chicago/Turabian StyleWestphal, Dominik Sebastian, Kathrin Pollmann, Christoph Marschall, Annette Wacker-Gussmann, Renate Oberhoffer-Fritz, Karl-Ludwig Laugwitz, Peter Ewert, and Cordula Maria Wolf. 2022. "It Is Not Carved in Stone—The Need for a Genetic Reevaluation of Variants in Pediatric Cardiomyopathies" Journal of Cardiovascular Development and Disease 9, no. 2: 41. https://doi.org/10.3390/jcdd9020041
APA StyleWestphal, D. S., Pollmann, K., Marschall, C., Wacker-Gussmann, A., Oberhoffer-Fritz, R., Laugwitz, K.-L., Ewert, P., & Wolf, C. M. (2022). It Is Not Carved in Stone—The Need for a Genetic Reevaluation of Variants in Pediatric Cardiomyopathies. Journal of Cardiovascular Development and Disease, 9(2), 41. https://doi.org/10.3390/jcdd9020041