
Further Evidence of Autosomal Recessive Inheritance of RPL3L Pathogenic Variants with Rapidly Progressive Neonatal Dilated Cardiomyopathy

 , and
, and 
Abstract
:1. Introduction
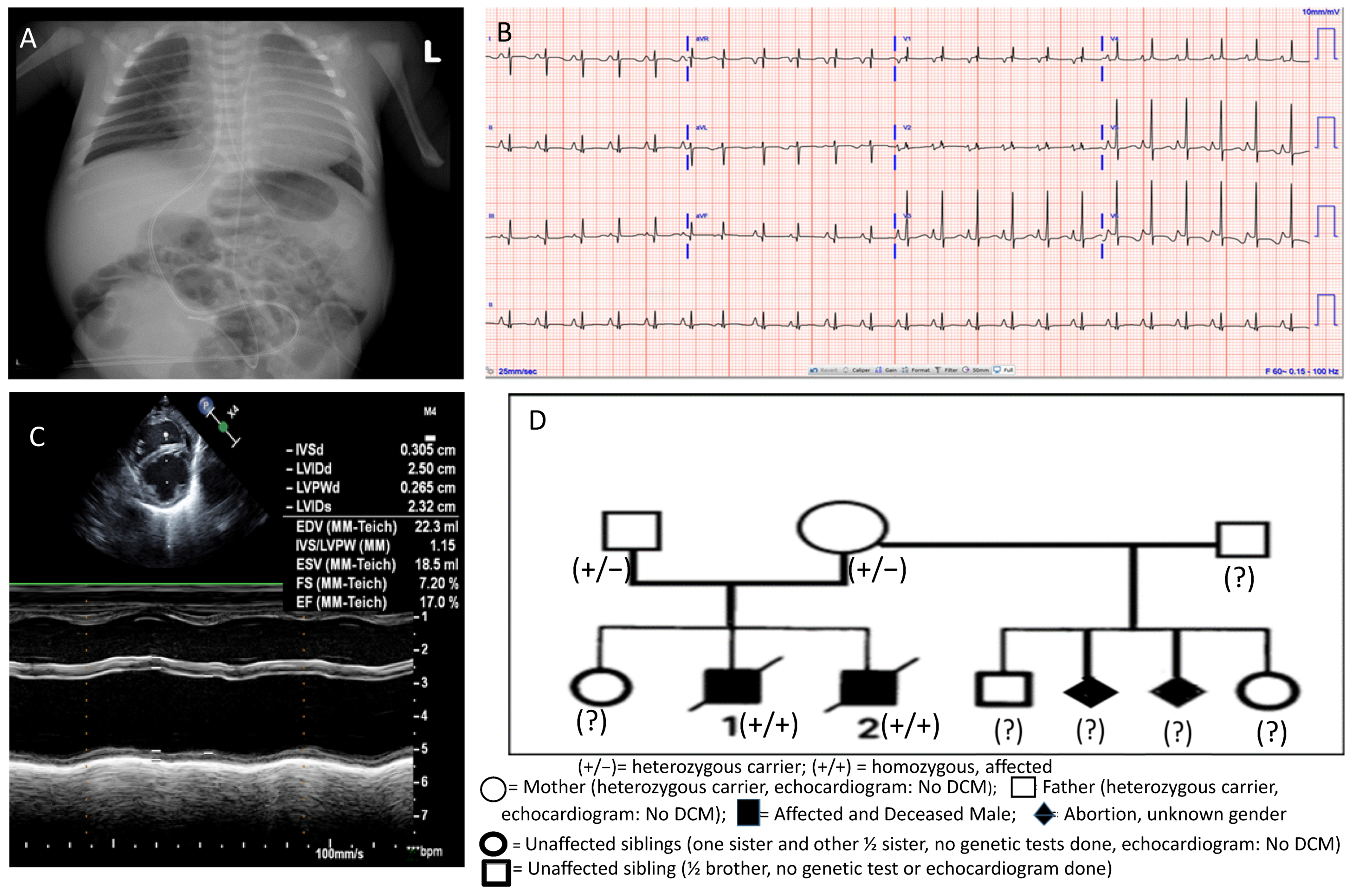
2. Case Presentation
3. Meta-Analysis of All Seven DCM Cases Due to RPL3L Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015, 16, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willig, L.K.; Petrikin, J.E.; Smith, L.D.; Saunders, C.J.; Thiffault, I.; Miller, N.A.; Soden, S.E.; Cakici, J.A.; Herd, S.M.; Twist, G.; et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: A retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 2015, 3, 377–387. [Google Scholar] [CrossRef] [Green Version]
- The NICUSeq Study Group. Effect of Whole-Genome sequencing on the clinical management of acutely ill infants with suspected genetic diseases. JAMA Pediatr. 2021, 175, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.-L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mut. 2015, 36, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, P.L.; Reiner, A.P.; Wang, G.; Kang, H.M.; Abecasis, G.R.; Altshuler, D.; Bamshad, M.J.; Nickerson, D.A.; Tracy, R.P.; Rich, S.S.; et al. Guidelines for large-scale sequence-based complex trait association studies: Lessons learned from the NHLBI Exosome Sequencing Project. Am. J. Hum. Genet. 2016, 99, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilescu, C.; Ojala, T.H.; Brilhante, V.; Ojanen, S.; Hinterding, H.M.; Palin, E.; Tero-Pekka Alastalo, T.-P.; Juha Koskenvuo, J.; Hiippala, A.; Jokinen, E.; et al. Genetic basis of severe childhood-onset cardiomyopathies. J. Am. Coll. Cardiol. 2018, 72, 2324–2338. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hassnan, Z.N.; Almesend, A.; Tulbah, S.; Alkakhgasg, A.; Alhadeq, F.; Alruwaili, N.; Alkorashy, M.; Alhashem, A.; Alrashdan, A.; Faqeih, E.; et al. Categorized genetic analysis in childhood-onset cardiomyopathy. Circ. Genomic Precis. Med. 2020, 13, e002969. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, M.; Argyriou, L.; Martínez-Azorín, F.; Morlot, S.; Yigit, G.; Lee, T.M.; Auber, B.; von Gise, A.; Petrey, D.S.; Thiele, H.; et al. Bi-allelic missense disease-causing variants in RPL3L associate neonatal dilated cardiomyopathy with muscle-specific ribosome biogenesis. Hum. Genet. 2020, 139, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaillou, T.; Zhang, X.; McCarthy, J.J. Expression of muscle-specific ribosomal protein L3-like impairs myotube growth. J. Cell. Physiol. 2016, 231, 1894–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Nielsen, J.B.; Jonsson, S.; Halldorsson, G.H.; Melsted, P.; Ivarsdottir, E.V.; Davidsson, O.B.; Kristjansson, R.P.; et al. Coding variants in RPL3L and MYZAP increase risk of atrial fibrillation. Commun. Biol. 2018, 1, 68. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.C.; Hall, A.W.; Choi, S.H.; Jurgens, S.J.; Haessler, J.; Bihlmeyer, N.A.; Grarup, N.; Lin, H.; Teumer, A.; Li-Gao, R.; et al. Genetic determinants of electrocardiographic P-wave duration and relation to atrial fibrillation. Circ. Genomic Precis. Med. 2020, 13, 387–395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Case No. | Sex | Consanguinity | Age at Diagn. of DCM | Country | Additional Cardiac Findings | Alive or Dead | Variants Identified | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | F | No | Day 1 | Germany | PFO PH | Died 21st DOL | c.923A > T (p.Asp308Val) and c.1027C > T (p.Arg343Trp) | [11] |
| 2 | M | No | Day 6 | Germany | TR | Died 15th DOL | c.923A > T (p.Asp308Val) and c.1027C > T (p.Arg343Trp) | [11] |
| 3 | M | Yes | Day 75 | Colombia | TR, MR RBBB ST-T abnormalities | HT at 6 months Alive at 9 years | c.566C > T (p.Thr189Met) and c.922G > A (p.Asp308Asn) | [11] |
| 4 | F | No | Day 48 | Spain | MR ST and T abnormalities | HT at 5 months Alive at 10 years | c.80G > A (p.Gly27Asp) and c.481C > T (p.Arg161Trp) | [11] |
| 5 | M | No | Day 12 | Spain | MR, TR ST and T abnormalities VSD | Died 30th DOL | c.80G > A (p.Gly27Asp) and c.481C > T (p.Arg161Trp) | [11] |
| 6 | M | No | Day 1 | USA | MR ST abnormalities | Died 124th DOL | c.1076_1080delCCGTG (p.Ala359Glyfs*4) and c.80G > A (p.Gly27Asp) | Present |
| 7 | M | No | Day 49 | USA | MR ST abnormalities | Died 56th DOL | c.1076_1080delCCGTG (p.Ala359Glyfs*4) and c.80G > A (p.Gly27Asp) | Present |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nannapaneni, H.; Ghaleb, S.; Arya, S.; Gajula, V.; Taylor, M.B.; Das, B.B. Further Evidence of Autosomal Recessive Inheritance of RPL3L Pathogenic Variants with Rapidly Progressive Neonatal Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2022, 9, 65. https://doi.org/10.3390/jcdd9030065
Nannapaneni H, Ghaleb S, Arya S, Gajula V, Taylor MB, Das BB. Further Evidence of Autosomal Recessive Inheritance of RPL3L Pathogenic Variants with Rapidly Progressive Neonatal Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease. 2022; 9(3):65. https://doi.org/10.3390/jcdd9030065
Chicago/Turabian StyleNannapaneni, Hemanth, Stephanie Ghaleb, Sandeep Arya, Viswanath Gajula, Mary B. Taylor, and Bibhuti B. Das. 2022. "Further Evidence of Autosomal Recessive Inheritance of RPL3L Pathogenic Variants with Rapidly Progressive Neonatal Dilated Cardiomyopathy" Journal of Cardiovascular Development and Disease 9, no. 3: 65. https://doi.org/10.3390/jcdd9030065
APA StyleNannapaneni, H., Ghaleb, S., Arya, S., Gajula, V., Taylor, M. B., & Das, B. B. (2022). Further Evidence of Autosomal Recessive Inheritance of RPL3L Pathogenic Variants with Rapidly Progressive Neonatal Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease, 9(3), 65. https://doi.org/10.3390/jcdd9030065