
The Role of POPDC Proteins in Cardiac Pacemaking and Conduction



Abstract
1. Introduction
2. Genomic Organization and Gene Regulation of POPDC Genes
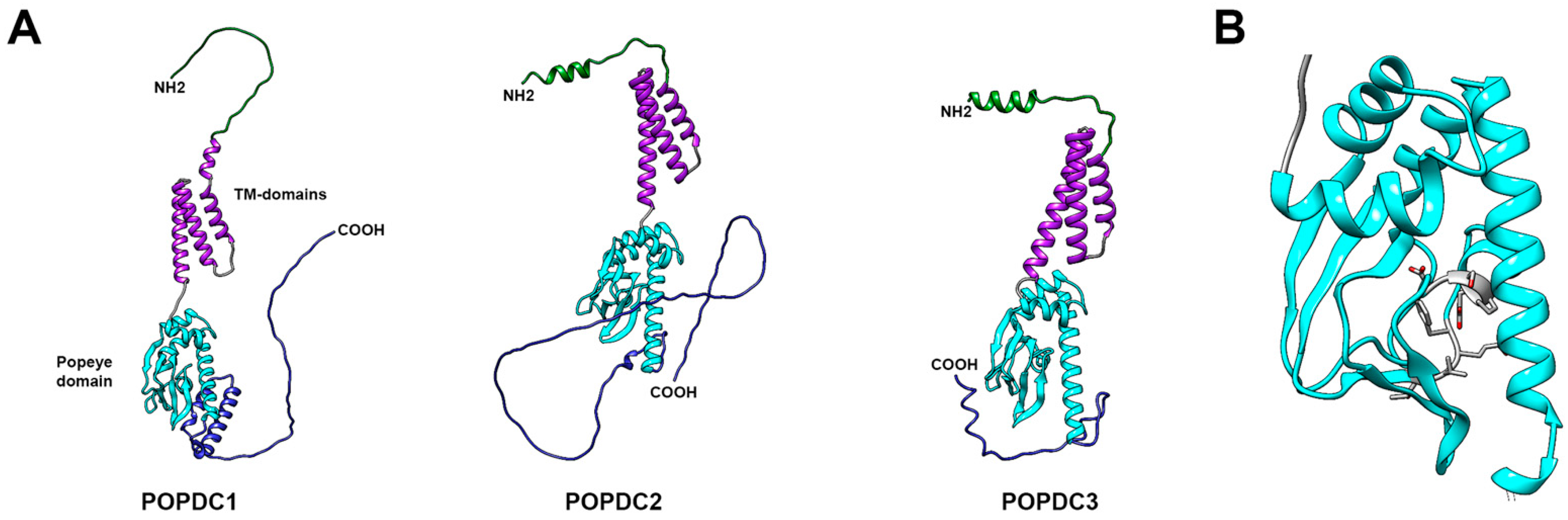
3. The POPDC Protein Family
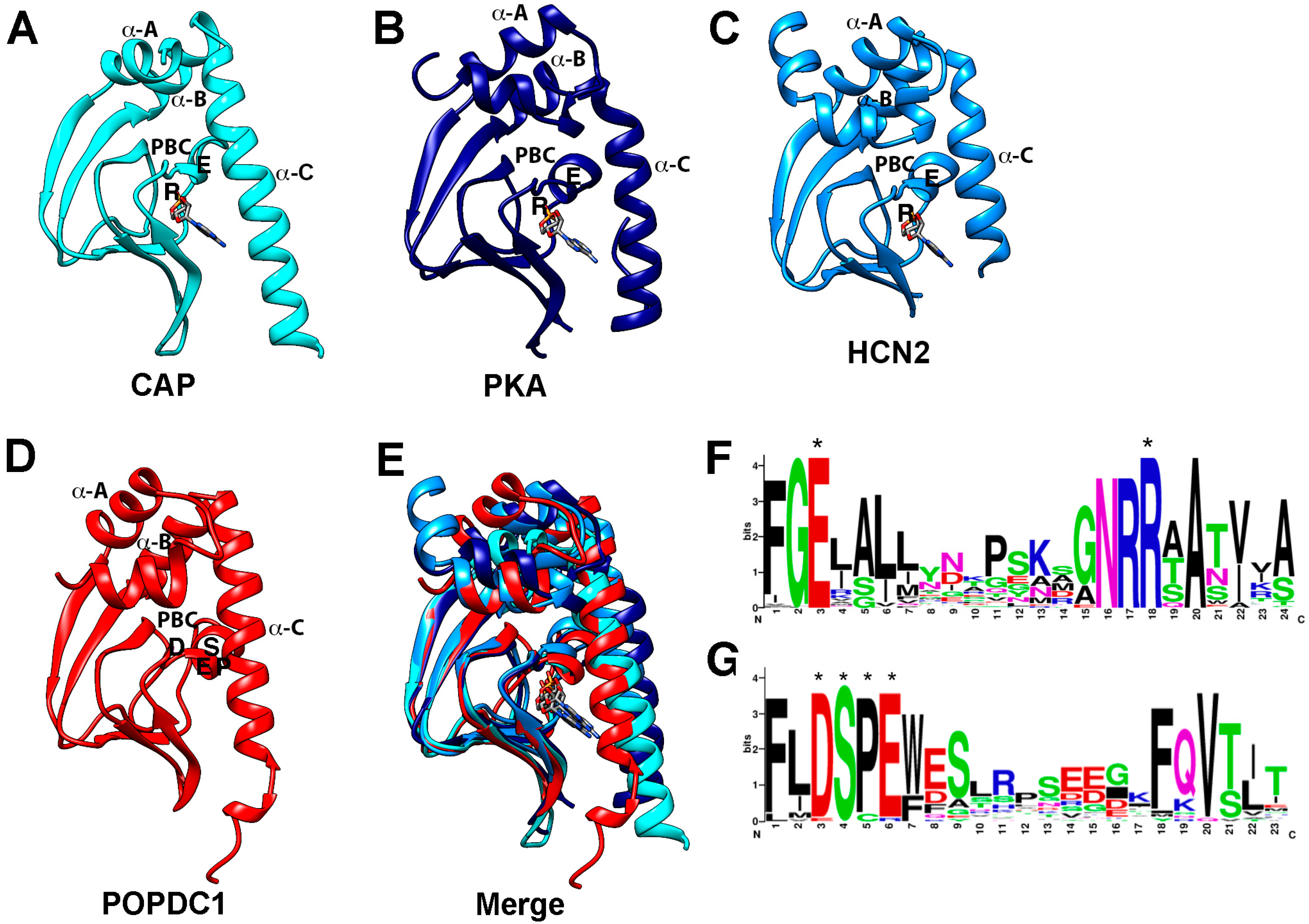
4. Elements of the cAMP Signaling Pathway
5. cAMP Effector Proteins
6. POPDC Proteins and Cardiac Arrhythmias
7. POPDC Proteins and Plasma Membrane Compartments
8. Mutations in POPDC Genes Are Causing Heart and Muscle Disease
9. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Lang, D.; Glukhov, A.V. Cellular and molecular mechanisms of functional hierarchy of pacemaker clusters in the sinoatrial node: New insights into sick sinus syndrome. J. Cardiovasc. Dev. Dis. 2021, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Choquet, C.; Boulgakoff, L.; Kelly, R.G.; Miquerol, L. New insights into the development and morphogenesis of the cardiac purkinje fiber network: Linking architecture and function. J. Cardiovasc. Dev. Dis. 2021, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Fedele, L.; Brand, T. The intrinsic cardiac nervous system and its role in cardiac pacemaking and conduction. J. Cardiovasc. Dev. Dis. 2020, 7, 54. [Google Scholar] [CrossRef]
- Andrée, B.; Hillemann, T.; Kessler-Icekson, G.; Schmitt-John, T.; Jockusch, H.; Arnold, H.H.; Brand, T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev. Biol. 2000, 223, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Reese, D.E.; Zavaljevski, M.; Streiff, N.L.; Bader, D. bves: A novel gene expressed during coronary blood vessel development. Dev. Biol. 1999, 209, 159–171. [Google Scholar] [CrossRef]
- Brand, T. The popeye domain-containing gene family. Cell Biochem. Biophys. 2005, 43, 95–104. [Google Scholar] [CrossRef]
- Gingold-Belfer, R.; Bergman, M.; Alcalay, Y.; Schlesinger, H.; Aravot, D.; Berman, M.; Salman, H.; Brand, T.; Kessler-Icekson, G. Popeye domain-containing 1is down-regulated in failing human hearts. Int. J. Mol. Med. 2010. [Google Scholar] [CrossRef]
- Gingold-Belfer, R.; Kessler-Icekson, G.; Morgenstern, S.; Rath-Wolfson, L.; Zemel, R.; Boltin, D.; Levi, Z.; Herman-Edelstein, M. The Transition from Gastric Intestinal Metaplasia to Gastric Cancer Involves POPDC1 and POPDC3 Downregulation. Int. J. Mol. Sci. 2021, 22, 5359. [Google Scholar] [CrossRef]
- Breher, S.S.; Mavridou, E.; Brenneis, C.; Froese, A.; Arnold, H.H.; Brand, T. Popeye domain containing gene 2 (Popdc2) is a myocyte-specific differentiation marker during chick heart development. Dev. Dyn. 2004, 229, 695–702. [Google Scholar] [CrossRef]
- Vasavada, T.K.; DiAngelo, J.R.; Duncan, M.K. Developmental expression of Pop1/Bves. J. Histochem. Cytochem. 2004, 52, 371–377. [Google Scholar] [CrossRef]
- Andrée, B.; Fleige, A.; Arnold, H.H.; Brand, T. Mouse Pop1 is required for muscle regeneration in adult skeletal muscle. Mol. Cell. Biol. 2002, 22, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Brand, T. Expression pattern of Popdc2 during mouse embryogenesis and in the adult. Dev. Dyn. 2008, 237, 780–787. [Google Scholar] [CrossRef]
- Ripley, A.N.; Chang, M.S.; Bader, D.M. Bves is expressed in the epithelial components of the retina, lens, and cornea. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2475–2483. [Google Scholar] [CrossRef]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinne, S.; Wischmeyer, E.; Schlueter, J.; Becher, J.; Simrick, S.; et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122, 1119–1130. [Google Scholar] [CrossRef]
- Barber, T.D.; Barber, M.C.; Tomescu, O.; Barr, F.G.; Ruben, S.; Friedman, T.B. Identification of target genes regulated by PAX3 and PAX3-FKHR in embryogenesis and alveolar rhabdomyosarcoma. Genomics 2002, 79, 278–284. [Google Scholar] [CrossRef]
- Han, P.; Fu, Y.; Liu, J.; Wang, Y.; He, J.; Gong, J.; Li, M.; Tan, Q.; Li, D.; Luo, Y.; et al. Netrin-1 promotes cell migration and invasion by down-regulation of BVES expression in human hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 1396–1409. [Google Scholar] [PubMed]
- Kim, M.; Jang, H.R.; Haam, K.; Kang, T.W.; Kim, J.H.; Kim, S.Y.; Noh, S.M.; Song, K.S.; Cho, J.S.; Jeong, H.Y.; et al. Frequent silencing of popeye domain-containing genes, BVES and POPDC3, is associated with promoter hypermethylation in gastric cancer. Carcinogenesis 2010, 31, 1685–1693. [Google Scholar] [CrossRef]
- Williams, C.S.; Zhang, B.; Smith, J.J.; Jayagopal, A.; Barrett, C.W.; Pino, C.; Russ, P.; Presley, S.H.; Peng, D.; Rosenblatt, D.O.; et al. BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J. Clin. Investig. 2011, 121, 4056–4069. [Google Scholar] [CrossRef]
- Amunjela, J.N.; Tucker, S.J. POPDC1 is suppressed in human breast cancer tissues and is negatively regulated by EGFR in breast cancer cell lines. Cancer Lett. 2017, 406, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhao, D.; Bownes, M. Blood vessel/epicardial substance (bves) expression, essential for embryonic development, is down regulated by Grk/EFGR signalling. Int. J. Dev. Biol. 2007, 51, 37–44. [Google Scholar] [CrossRef]
- Parnes, D.; Jacoby, V.; Sharabi, A.; Schlesinger, H.; Brand, T.; Kessler-Icekson, G. The Popdc gene family in the rat: Molecular cloning, characterization and expression analysis in the heart and cultured cardiomyocytes. Biochim. Biophys. Acta 2007, 1769, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Dupays, L.; Shang, C.; Wilson, R.; Kotecha, S.; Wood, S.; Towers, N.; Mohun, T. Sequential Binding of MEIS1 and NKX2-5 on the Popdc2 Gene: A Mechanism for Spatiotemporal Regulation of Enhancers during Cardiogenesis. Cell Rep. 2015, 13, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Pezhouman, A.; Engel, J.L.; Nguyen, N.B.; Skelton, R.J.P.; Gilmore, W.B.; Qiao, R.; Sahoo, D.; Zhao, P.; Elliott, D.A.; Ardehali, R. Isolation and characterization of hESC-derived heart field-specific cardiomyocytes unravels new insights into their transcriptional and electrophysiological profiles. Cardiovasc. Res. 2021, in press. [Google Scholar] [CrossRef]
- Alcalay, Y.; Hochhauser, E.; Kliminski, V.; Dick, J.; Zahalka, M.A.; Parnes, D.; Schlesinger, H.; Abassi, Z.; Shainberg, A.; Schindler, R.F.; et al. Popeye domain containing 1 (popdc1/bves) is a caveolae-associated protein involved in ischemia tolerance. PLoS ONE 2013, 8, e71100. [Google Scholar] [CrossRef]
- Wada, A.M.; Reese, D.E.; Bader, D.M. Bves: Prototype of a new class of cell adhesion molecules expressed during coronary artery development. Development 2001, 128, 2085–2093. [Google Scholar] [CrossRef]
- Knight, R.F.; Bader, D.M.; Backstrom, J.R. Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J. Biol. Chem. 2003, 278, 32872–32879. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Swan, A.H.; Gruscheski, L.; Boland, L.A.; Brand, T. The Popeye domain containing gene family encoding a family of cAMP-effector proteins with important functions in striated muscle and beyond. J. Muscle Res. Cell Motil. 2019, 40, 169–183. [Google Scholar] [CrossRef]
- Schindler, R.F.; Scotton, C.; Zhang, J.; Passarelli, C.; Ortiz-Bonnin, B.; Simrick, S.; Schwerte, T.; Poon, K.L.; Fang, M.; Rinne, S.; et al. POPDC1S201F causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Investig. 2016, 126, 239–253. [Google Scholar] [CrossRef]
- Rodrigues, J.G.; Balmana, M.; Macedo, J.A.; Pocas, J.; Fernandes, A.; de-Freitas-Junior, J.C.M.; Pinho, S.S.; Gomes, J.; Magalhaes, A.; Gomes, C.; et al. Glycosylation in cancer: Selected roles in tumour progression, immune modulation and metastasis. Cell. Immunol. 2018, 333, 46–57. [Google Scholar] [CrossRef]
- Moore, C.J.; Hewitt, J.E. Dystroglycan glycosylation and muscular dystrophy. Glycoconj. J. 2009, 26, 349–357. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Hager, H.A.; Wada, A.; Koyama, T.; Chang, M.S.; Bader, D.M. Identification of a novel intracellular interaction domain essential for Bves function. PLoS ONE 2008, 3, e2261. [Google Scholar] [CrossRef]
- Russ, P.K.; Pino, C.J.; Williams, C.S.; Bader, D.M.; Haselto, F.R.; Chang, M.S. Bves modulates tight junction associated signaling. PLoS ONE 2011, 6, e14563. [Google Scholar] [CrossRef] [PubMed]
- Busby, S.; Ebright, R.H. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 1999, 293, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, G.S.; Korfali, N.; Swanson, S.K.; Malik, P.; Srsen, V.; Batrakou, D.G.; de las Heras, J.; Zuleger, N.; Kerr, A.R.; Florens, L.; et al. Several novel nuclear envelope transmembrane proteins identified in skeletal muscle have cytoskeletal associations. Mol. Cell. Proteom. 2011, 10, M110-003129. [Google Scholar] [CrossRef]
- Schindler, R.; Simrick, S.; Brand, T. Nuclear localization of members of popeye domain containing (Popdc) protein family. Cardiovasc. Res. 2012, 93, S98. [Google Scholar]
- Brand, T. The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle. J. Cardiovasc. Dev. Dis. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Tan, N.; Chung, M.K.; Smith, J.D.; Hsu, J.; Serre, D.; Newton, D.W.; Castel, L.; Soltesz, E.; Pettersson, G.; Gillinov, A.M.; et al. Weighted gene coexpression network analysis of human left atrial tissue identifies gene modules associated with atrial fibrillation. Circ. Cardiovasc. Genet. 2013, 6, 362–371. [Google Scholar] [CrossRef]
- Lundby, A.; Andersen, M.N.; Steffensen, A.B.; Horn, H.; Kelstrup, C.D.; Francavilla, C.; Jensen, L.J.; Schmitt, N.; Thomsen, M.B.; Olsen, J.V. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci. Signal. 2013, 6, rs11. [Google Scholar] [CrossRef]
- Hager, H.A.; Roberts, R.J.; Cross, E.E.; Proux-Gillardeaux, V.; Bader, D.M. Identification of a novel Bves function: Regulation of vesicular transport. EMBO J. 2010, 29, 532–545. [Google Scholar] [CrossRef]
- Osler, M.E.; Chang, M.S.; Bader, D.M. Bves modulates epithelial integrity through an interaction at the tight junction. J. Cell Sci. 2005, 118, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Hager, H.A.; Francis, R.; Kilkenny, D.M.; Lo, C.W.; Bader, D.M. Bves directly interacts with GEFT, and controls cell shape and movement through regulation of Rac1/Cdc42 activity. Proc. Natl. Acad. Sci. USA 2008, 105, 8298–8303. [Google Scholar] [CrossRef]
- Schindler, R.F.; Scotton, C.; French, V.; Ferlini, A.; Brand, T. The Popeye Domain Containing Genes and their Function in Striated Muscle. J. Cardiovasc. Dev. Dis. 2016, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.; Fuller, H.R.; Schindler, R.F.R.; Shirran, S.L.; Brand, T.; Morris, G.E. An interaction of heart disease-associated proteins POPDC1/2 with XIRP1 in transverse tubules and intercalated discs. BMC Mol. Cell. Biol. 2020, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Berthet, J.; Rall, T.W.; Sutherland, E.W. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J. Biol. Chem. 1957, 224, 463–475. [Google Scholar]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef]
- Kamenetsky, M.; Middelhaufe, S.; Bank, E.M.; Levin, L.R.; Buck, J.; Steegborn, C. Molecular details of cAMP generation in mammalian cells: A tale of two systems. J. Mol. Biol. 2006, 362, 623–639. [Google Scholar] [CrossRef]
- El-Armouche, A.; Eschenhagen, T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 2009, 14, 225–241. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef]
- Hong, T.; Shaw, R.M. Cardiac T-Tubule Microanatomy and Function. Physiol. Rev. 2017, 97, 227–252. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bunemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; Clancy, C.E.; Harvey, R.D. Mechanisms Restricting Diffusion of Intracellular cAMP. Sci. Rep. 2016, 6, 19577. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in.n.subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.A.; Dessauer, C.W. Function of Adenylyl Cyclase in Heart: The AKAP Connection. J. Cardiovasc. Dev. Dis. 2018, 5, 2. [Google Scholar] [CrossRef]
- Brust, T.F. Adenylyl Cyclases. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Mattick, P.; Parrington, J.; Odia, E.; Simpson, A.; Collins, T.; Terrar, D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J. Physiol. 2007, 582, 1195–1203. [Google Scholar] [CrossRef]
- Younes, A.; Lyashkov, A.E.; Graham, D.; Sheydina, A.; Volkova, M.V.; Mitsak, M.; Vinogradova, T.M.; Lukyanenko, Y.O.; Li, Y.; Ruknudin, A.M.; et al. Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 2008, 283, 14461–14468. [Google Scholar] [CrossRef]
- Moen, J.M.; Matt, M.G.; Ramirez, C.; Tarasov, K.V.; Chakir, K.; Tarasova, Y.S.; Lukyanenko, Y.; Tsutsui, K.; Monfredi, O.; Morrell, C.H.; et al. Overexpression of a Neuronal Type Adenylyl Cyclase (Type 8) in Sinoatrial Node Markedly Impacts Heart Rate and Rhythm. Front. Neurosci. 2019, 13, 615. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.L.; Jacobson, K.L.; Singh, J.C.; Idzerda, R.; Ott, S.M.; DiJulio, D.H.; Wong, S.T.; Storm, D.R. The type 8 adenylyl cyclase is critical for Ca2+ stimulation of cAMP accumulation in mouse parotid acini. J. Biol. Chem. 2000, 275, 14691–14699. [Google Scholar] [CrossRef]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef]
- Molenaar, P.; Christ, T.; Hussain, R.I.; Engel, A.; Berk, E.; Gillette, K.T.; Chen, L.; Galindo-Tovar, A.; Krobert, K.A.; Ravens, U.; et al. PDE3, but not PDE4, reduces beta(1)- and beta(2)-adrenoceptor-mediated inotropic and lusitropic effects in failing ventricle from metoprolol-treated patients. Br. J. Pharmacol. 2013, 169, 528–538. [Google Scholar] [CrossRef]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Kaumann, A.J. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (−)-adrenaline mediated through mouse cardiac beta(1)-adrenoceptors. Br. J. Pharmacol. 2008, 153, 710–720. [Google Scholar] [CrossRef]
- Tibbo, A.J.; Baillie, G.S. Phosphodiesterase 4B: Master Regulator of Brain Signaling. Cells 2020, 9, 1254. [Google Scholar] [CrossRef]
- Taylor, S.S.; Ilouz, R.; Zhang, P.; Kornev, A.P. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.H.; Scott, J.D. AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol. Sci. 2020, 41, 933–946. [Google Scholar] [CrossRef]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar] [CrossRef]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Rehmann, H.; Lao, D.H.; Erickson, J.R.; Bossuyt, J.; Chen, J.; Bers, D.M. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 3991–3996. [Google Scholar] [CrossRef]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional Mitochondrial Epac1 Controls Myocardial Cell Death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef]
- Brette, F.; Blandin, E.; Simard, C.; Guinamard, R.; Salle, L. Epac activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle. J. Mol. Cell Cardiol. 2013, 57, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Layh, B.; Ludwig, A. Novel insights into the distribution of cardiac HCN channels: An expression study in the mouse heart. J. Mol. Cell Cardiol. 2011, 51, 997–1006. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Biel, M. HCN channels: Structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 2009, 66, 470–494. [Google Scholar] [CrossRef]
- Fenske, S.; Hennis, K.; Rotzer, R.D.; Brox, V.F.; Becirovic, E.; Scharr, A.; Gruner, C.; Ziegler, T.; Mehlfeld, V.; Brennan, J.; et al. cAMP-dependent regulation of HCN4 controls the tonic entrainment process in sinoatrial node pacemaker cells. Nat. Commun. 2020, 11, 5555. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef]
- Tsutsui, K.; Monfredi, O.J.; Sirenko-Tagirova, S.G.; Maltseva, L.A.; Bychkov, R.; Kim, M.S.; Ziman, B.D.; Tarasov, K.V.; Tarasova, Y.S.; Zhang, J.; et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal. 2018, 11, eaap7608. [Google Scholar] [CrossRef]
- Tsutsui, K.; Florio, M.C.; Yang, A.; Wirth, A.N.; Yang, D.; Kim, M.S.; Ziman, B.D.; Bychkov, R.; Monfredi, O.J.; Maltsev, V.A.; et al. cAMP-Dependent Signaling Restores AP Firing in Dormant SA Node Cells via Enhancement of Surface Membrane Currents and Calcium Coupling. Front. Physiol. 2021, 12, 596832. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef]
- Fenske, S.; Krause, S.C.; Hassan, S.I.; Becirovic, E.; Auer, F.; Bernard, R.; Kupatt, C.; Lange, P.; Ziegler, T.; Wotjak, C.T.; et al. Sick sinus syndrome in HCN1-deficient mice. Circulation 2013, 128, 2585–2594. [Google Scholar] [CrossRef]
- Sargento, L.; Satendra, M.; Longo, S.; Lousada, N.; Dos Reis, R.P. Heart Rate Reduction with Ivabradine in Patients with Acute Decompensated Systolic Heart Failure. Am. J. Cardiovasc. Drugs 2014, 14, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Hennis, K.; Biel, M.; Wahl-Schott, C.; Fenske, S. Beyond pacemaking: HCN channels in sinoatrial node function. Prog. Biophys. Mol. Biol. 2021, 166, 51–60. [Google Scholar] [CrossRef]
- Alig, J.; Marger, L.; Mesirca, P.; Ehmke, H.; Mangoni, M.E.; Isbrandt, D. Control of heart rate by cAMP sensitivity of HCN channels. Proc. Natl. Acad. Sci. USA 2009, 106, 12189–12194. [Google Scholar] [CrossRef]
- Kozasa, Y.; Nakashima, N.; Ito, M.; Ishikawa, T.; Kimoto, H.; Ushijima, K.; Makita, N.; Takano, M. HCN4 pacemaker channels attenuate the parasympathetic response and stabilize the spontaneous firing of the sinoatrial node. J. Physiol. 2018, 596, 809–825. [Google Scholar] [CrossRef]
- Herrmann, S.; Stieber, J.; Stockl, G.; Hofmann, F.; Ludwig, A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007, 26, 4423–4432. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Fenske, S.; Biel, M. HCN channels: New roles in sinoatrial node function. Curr. Opin. Pharmacol. 2014, 15, 83–90. [Google Scholar] [CrossRef]
- Fenske, S.; Probstle, R.; Auer, F.; Hassan, S.; Marks, V.; Pauza, D.H.; Biel, M.; Wahl-Schott, C. Comprehensive multilevel in vivo and in vitro analysis of heart rate fluctuations in mice by ECG telemetry and electrophysiology. Nat. Protoc. 2016, 11, 61–86. [Google Scholar] [CrossRef]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Ten Eyck, L.F.; Goodsell, D.S.; Haste, N.M.; Kornev, A.; Taylor, S.S. The cAMP binding domain: An ancient signaling module. Proc. Natl. Acad. Sci. USA 2005, 102, 45–50. [Google Scholar] [CrossRef]
- Kannan, N.; Wu, J.; Anand, G.S.; Yooseph, S.; Neuwald, A.F.; Venter, J.C.; Taylor, S.S. Evolution of allostery in the cyclic nucleotide binding module. Genome Biol. 2007, 8, R264. [Google Scholar] [CrossRef] [PubMed]
- Torlopp, A.; Breher, S.S.; Schluter, J.; Brand, T. Comparative analysis of mRNA and protein expression of Popdc1 (Bves) during early development in the chick embryo. Dev. Dyn. 2006, 235, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Verheijck, E.E.; Wessels, A.; van Ginneken, A.C.; Bourier, J.; Markman, M.W.; Vermeulen, J.L.; de Bakker, J.M.; Lamers, W.H.; Opthof, T.; Bouman, L.N. Distribution of atrial and nodal cells within the rabbit sinoatrial node: Models of sinoatrial transition. Circulation 1998, 97, 1623–1631. [Google Scholar] [CrossRef]
- Wu, J.; Schuessler, R.B.; Rodefeld, M.D.; Saffitz, J.E.; Boineau, J.P. Morphological and membrane characteristics of spider and spindle cells isolated from rabbit sinus node. Am. J. Physiol. 2001, 280, H1232–H1240. [Google Scholar] [CrossRef]
- Kirchmaier, B.C.; Poon, K.L.; Schwerte, T.; Huisken, J.; Winkler, C.; Jungblut, B.; Stainier, D.Y.; Brand, T. The Popeye domain containing 2 (popdc2) gene in zebrafish is required for heart and skeletal muscle development. Dev. Biol. 2012, 363, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Malbouyres, M.; Pagnon-Minot, A.; Ruggiero, F.; Le Guellec, D. Development of the zebrafish myoseptum with emphasis on the myotendinous junction. Cell Tissue Res. 2011, 346, 439–449. [Google Scholar] [CrossRef]
- Goody, M.F.; Sher, R.B.; Henry, C.A. Hanging on for the ride: Adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev. Biol. 2015, 401, 75–91. [Google Scholar] [CrossRef]
- Choudhury, M.; Boyett, M.R.; Morris, G.M. Biology of the Sinus Node and its Disease. Arrhythm. Electrophysiol. Rev. 2015, 4, 28–34. [Google Scholar] [CrossRef]
- Simrick, S.; Schindler, R.F.; Poon, K.L.; Brand, T. Popeye domain-containing proteins and stress-mediated modulation of cardiac pacemaking. Trends Cardiovasc. Med. 2013, 23, 257–263. [Google Scholar] [CrossRef]
- Lubelwana Hafver, T.; Wanichawan, P.; Manfra, O.; de Souza, G.A.; Lunde, M.; Martinsen, M.; Louch, W.E.; Mathias Sejersted, O.; Carlson, C.R. Mapping the in vitro interactome of cardiac sodium (Na+)-calcium (Ca2+) exchanger 1 (NCX1). Proteomics 2017, 17, 1600417. [Google Scholar] [CrossRef] [PubMed]
- Torrente, A.G.; Zhang, R.; Zaini, A.; Giani, J.F.; Kang, J.; Lamp, S.T.; Philipson, K.D.; Goldhaber, J.I. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9769–9774. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Lakatta, E.G.; Maltsev, V.A. From two competing oscillators to one coupled-clock pacemaker cell system. Front. Physiol. 2015, 6, 28. [Google Scholar] [CrossRef]
- Decher, N.; Kiper, A.K.; Rinne, S. Stretch-activated potassium currents in the heart: Focus on TREK-1 and arrhythmias. Prog. Biophys. Mol. Biol. 2017, 130, 223–232. [Google Scholar] [CrossRef]
- Unudurthi, S.D.; Wu, X.; Qian, L.; Amari, F.; Onal, B.; Li, N.; Makara, M.A.; Smith, S.A.; Snyder, J.; Fedorov, V.V.; et al. Two-Pore K+ Channel TREK-1 Regulates Sinoatrial Node Membrane Excitability. J. Am. Heart Assoc. 2016, 5, e002865. [Google Scholar] [CrossRef]
- Tibbo, A.J.; Dobi, S.; McFall, A.; Tejeda, G.S.; Blair, C.; MacLeod, R.; MacQuaide, N.; Gök, C.; Fuller, W.; Smith, B.O.; et al. Phosphodiesterase Type 4 anchoring regulates cAMP signaling to Popeye domain-containing proteins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Terrenoire, C.; Lauritzen, I.; Lesage, F.; Romey, G.; Lazdunski, M. A TREK-1-like potassium channel in atrial cells inhibited by beta-adrenergic stimulation and activated by volatile anesthetics. Circ. Res. 2001, 89, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honore, E.; Maingret, F.; Lesage, F.; Fink, M.; Duprat, F.; Lazdunski, M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998, 17, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
- Sandoz, G.; Thummler, S.; Duprat, F.; Feliciangeli, S.; Vinh, J.; Escoubas, P.; Guy, N.; Lazdunski, M.; Lesage, F. AKAP150, a switch to convert mechano-, pH- and arachidonic acid-sensitive TREK K(+) channels into open leak channels. EMBO J. 2006, 25, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Shetty, M.S.; Ris, L.; Schindler, R.F.R.; Mizuno, K.; Fedele, L.; Giese, K.P.; Brand, T.; Abel, T. Mice lacking the cAMP effector protein POPDC1 show enhanced hippocampal synaptic plasticity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Abel, T.; Martin, K.C.; Bartsch, D.; Kandel, E.R. Memory suppressor genes: Inhibitory constraints on the storage of long-term memory. Science 1998, 279, 338–341. [Google Scholar] [CrossRef]
- Abel, T.; Nguyen, P.V.; Barad, M.; Deuel, T.A.; Kandel, E.R.; Bourtchouladze, R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 1997, 88, 615–626. [Google Scholar] [CrossRef]
- Blitzer, R.D.; Wong, T.; Nouranifar, R.; Iyengar, R.; Landau, E.M. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron 1995, 15, 1403–1414. [Google Scholar] [CrossRef]
- Bastiani, M.; Parton, R.G. Caveolae at a glance. J. Cell Sci. 2010, 123, 3831–3836. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Reilly, L.; Eckhardt, L.L. Caveolin-3 Microdomain: Arrhythmia Implications for Potassium Inward Rectifier and Cardiac Sodium Channel. Front. Physiol. 2018, 9, 1548. [Google Scholar] [CrossRef]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, ion channels and cardiac arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef]
- Harvey, R.D.; Calaghan, S.C. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J. Mol. Cell. Cardiol. 2012, 52, 366–375. [Google Scholar] [CrossRef]
- Soni, S.; Raaijmakers, A.J.; Raaijmakers, L.M.; Damen, J.M.; van Stuijvenberg, L.; Vos, M.A.; Heck, A.J.; van Veen, T.A.; Scholten, A. A Proteomics Approach to Identify New Putative Cardiac Intercalated Disk Proteins. PLoS ONE 2016, 11, e0152231. [Google Scholar] [CrossRef]
- Leo-Macias, A.; Liang, F.X.; Delmar, M. Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovasc. Res. 2015, 107, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, T.L.; Adams, E.; Lin, J.L.; Lin, J.J. Intercalated disc protein, mXinalpha, suppresses p120-catenin-induced branching phenotype via its interactions with p120-catenin and cortactin. Arch. Biochem. Biophys. 2013, 535, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Hu, S.; Wei, Y. Cardiomyopathy-Associated Gene 1-Sensitive PKC-Dependent Connexin 43 Expression and Phosphorylation in Left Ventricular Noncompaction Cardiomyopathy. Cell Physiol. Biochem. 2017, 44, 828–842. [Google Scholar] [CrossRef]
- Huang, L.; Wu, K.H.; Zhang, L.; Wang, Q.; Tang, S.; Wu, Q.; Jiang, P.H.; Lin, J.J.; Guo, J.; Wang, L.; et al. Critical Roles of Xirp Proteins in Cardiac Conduction and Their Rare Variants Identified in Sudden Unexplained Nocturnal Death Syndrome and Brugada Syndrome in Chinese Han Population. J. Am. Heart Assoc. 2018, 7, e006320. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [CrossRef]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. Elife 2013, 2, e00926. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Al-Sajee, D.; Nissar, A.A.; Coleman, S.K.; Rebalka, I.A.; Chiang, A.; Wathra, R.; van der Ven, P.F.; Orfanos, Z.; Hawke, T.J. Xin-deficient mice display myopathy, impaired contractility, attenuated muscle repair and altered satellite cell functionality. Acta Physiol. 2015, 214, 248–260. [Google Scholar] [CrossRef]
- Li, H.; Xu, L.; Gao, Y.; Zuo, Y.; Yang, Z.; Zhao, L.; Chen, Z.; Guo, J.S.; Han, R. BVES Is a Novel Interactor of ANO5 and Regulates Myoblast Differentiation. Res. Square 2021. [Google Scholar] [CrossRef]
- Foltz, S.J.; Cui, Y.Y.; Choo, H.J.; Hartzell, H.C. ANO5 ensures trafficking of annexins in wounded myofibers. J. Cell Biol. 2021, 220, e202007059. [Google Scholar] [CrossRef]
- De Ridder, W.; Nelson, I.; Asselbergh, B.; De Paepe, B.; Beuvin, M.; Ben Yaou, R.; Masson, C.; Boland, A.; Deleuze, J.F.; Maisonobe, T.; et al. Muscular dystrophy with arrhythmia caused by loss-of-function mutations in BVES. Neurol. Genet. 2019, 5, e321. [Google Scholar] [CrossRef]
- Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de Las Heras, J.I.; Dixon, C.R.; Harris, E.; Kolbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. EBioMedicine 2020, 51, 102587. [Google Scholar] [CrossRef]
- Beecher, G.; Tang, C.; Liewluck, T. Severe adolescent-onset limb-girdle muscular dystrophy due to a novel homozygous nonsense BVES variant. J. Neurol. Sci. 2021, 420, 117259. [Google Scholar] [CrossRef] [PubMed]
- Indrawati, L.A.; Iida, A.; Tanaka, Y.; Honma, Y.; Mizoguchi, K.; Yamaguchi, T.; Ikawa, M.; Hayashi, S.; Noguchi, S.; Nishino, I. Two Japanese LGMDR25 patients with a biallelic recurrent nonsense variant of BVES. Neuromuscul. Disord. 2020, 30, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.; Ortiz-Bonnin, B.; Stallmeyer, B.; Kiper, A.K.; Fortmuller, L.; Schindler, R.F.R.; Herbort-Brand, U.; Kabir, N.S.; Dittmann, S.; Friedrich, C.; et al. POPDC2 a novel susceptibility gene for conduction disorders. J. Mol. Cell. Cardiol. 2020, 145, 74–83. [Google Scholar] [CrossRef]
- Helm, B.; Swan, A.H..; Brand, T.; Steinberg, L.A.; Kean, A.C. Using exome sequencing to uncover a POPDC2 variant as a novel candidate cause of familial junctional ectopic tachycardia. Heart Rhythm. 2021, 18, S381. [Google Scholar] [CrossRef]
- Vissing, J.; Johnson, K.; Topf, A.; Nafissi, S.; Diaz-Manera, J.; French, V.M.; Schindler, R.F.; Sarathchandra, P.; Lokken, N.; Rinne, S.; et al. POPDC3 Gene Variants Associate with a New Form of Limb Girdle Muscular Dystrophy. Ann. Neurol. 2019, 86, 832–843. [Google Scholar] [CrossRef]
- Barton, E.R.; Pacak, C.A.; Stoppel, W.L.; Kang, P.B. The ties that bind: Functional clusters in limb-girdle muscular dystrophy. Skelet. Muscle 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Puckelwartz, M.; McNally, E.M. Emery-Dreifuss muscular dystrophy. Handb. Clin. Neurol. 2011, 101, 155–166. [Google Scholar] [CrossRef]
- Wang, X.; Tucker, N.R.; Rizki, G.; Mills, R.; Krijger, P.H.; de Wit, E.; Subramanian, V.; Bartell, E.; Nguyen, X.X.; Ye, J.; et al. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife 2016, 5, e10557. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Giraldez, R.; Gogarten, S.M.; Below, J.E.; Yao, J.; Seyerle, A.A.; Highland, H.M.; Kooperberg, C.; Soliman, E.Z.; Rotter, J.I.; Kerr, K.F.; et al. GWAS of the electrocardiographic QT interval in Hispanics/Latinos generalizes previously identified loci and identifies population-specific signals. Sci. Rep. 2017, 7, 17075. [Google Scholar] [CrossRef]
- Xi, Y.; Honeywell, C.; Zhang, D.; Schwartzentruber, J.; Beaulieu, C.L.; Tetreault, M.; Hartley, T.; Marton, J.; Vidal, S.M.; Majewski, J.; et al. Whole exome sequencing identifies the TNNI3K gene as a cause of familial conduction system disease and congenital junctional ectopic tachycardia. Int. J. Cardiol. 2015, 185, 114–116. [Google Scholar] [CrossRef]
- Hund, T.J.; Koval, O.M.; Li, J.; Wright, P.J.; Qian, L.; Snyder, J.S.; Gudmundsson, H.; Kline, C.F.; Davidson, N.P.; Cardona, N.; et al. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Investig. 2010, 120, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Anderson, M.E. CaMKII in sinoatrial node physiology and dysfunction. Front. Pharmacol. 2014, 5, 48. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Mutation | Trait | Phenotype | Ref. |
|---|---|---|---|---|
| POPDC1 | 1A>G | rec. | AV-block, Myalgia, hCK 1 | [134] |
| R88X | rec. | AV-block, LGMD 2, hCK | [134] | |
| D92G | rec. | EDMD 3 | [135] | |
| R143X | rec. | AV-block, LGMD, hCK | [136] | |
| S201F 4 | rec. | AV-block, LGMD, hCK | [29] | |
| Del56V217-K272 | rec. | AV-block, LGMD, hCK | [134] | |
| S263X | rec. | AV-block, LGMD, hCK | [137] | |
| POPDC2 | W188X | dom. | AV-block | [138] |
| L245P | dom. | cJET 5 | [139] | |
| POPDC3 | L155H | rec. | LGMD | [140] |
| L217F | rec. | LGMD | [140] | |
| R261Q | rec. | LGMD | [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruscheski, L.; Brand, T. The Role of POPDC Proteins in Cardiac Pacemaking and Conduction. J. Cardiovasc. Dev. Dis. 2021, 8, 160. https://doi.org/10.3390/jcdd8120160
Gruscheski L, Brand T. The Role of POPDC Proteins in Cardiac Pacemaking and Conduction. Journal of Cardiovascular Development and Disease. 2021; 8(12):160. https://doi.org/10.3390/jcdd8120160
Chicago/Turabian StyleGruscheski, Lena, and Thomas Brand. 2021. "The Role of POPDC Proteins in Cardiac Pacemaking and Conduction" Journal of Cardiovascular Development and Disease 8, no. 12: 160. https://doi.org/10.3390/jcdd8120160
APA StyleGruscheski, L., & Brand, T. (2021). The Role of POPDC Proteins in Cardiac Pacemaking and Conduction. Journal of Cardiovascular Development and Disease, 8(12), 160. https://doi.org/10.3390/jcdd8120160