Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of KATP Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Ventricular Fibroblast Isolation
2.3. Characteristics of pH, PO2 and PCO2 During Ischemia and Reperfusion
2.4. Effect of Ischemia Reperfusion Injury on Fibroblast to Myofibroblast Differentiation
2.5. Effect of Ischemic Preconditioning on Ischemia Reperfusion Injury Induced Fibroblast to Myofibroblast Differentiation
2.6. Role of Adenosine Triphosphate-Sensitive Potassium Channels in Ischemic Preconditioning
2.7. Immunostaining for α-Smooth Muscle Actin
2.8. Data Analysis and Statistics
2.8.1. Characterization of the Ischemic Conditions
2.8.2. Ischemia Reperfusion Injury and Ischemic Preconditioning
3. Results
3.1. Characteristics of Conditions During Ischemia and Reperfusion
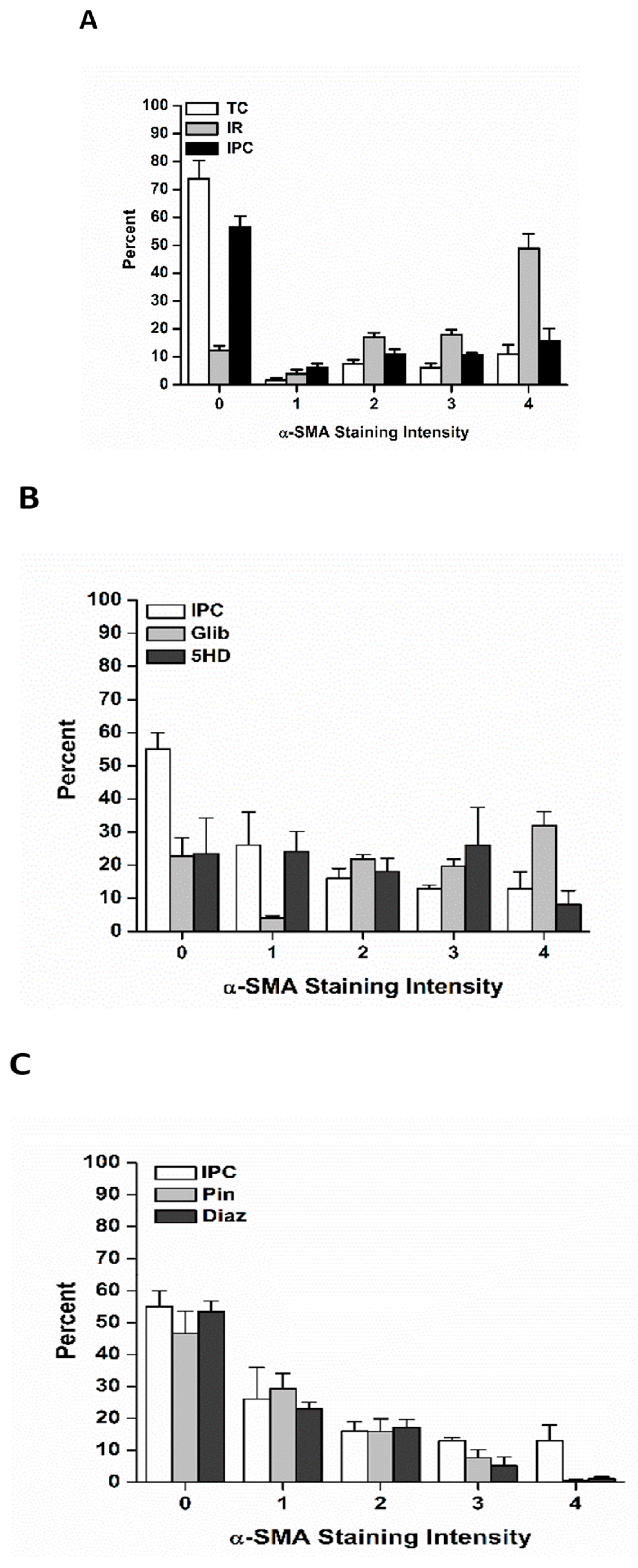
3.2. The Effects of Ischemia Reperfusion on Cardiac Fibroblast Differentiation
3.3. The Effects of Ischemic Preconditioning on Cardiac Fibroblast Differentiation, and the Role of Adenosine Triphosphate-Sensitive Potassium Channels
3.3.1. The effects of Pinacidil and Diazoxide Treatment on Ischemia Reperfusion-Induced Cardiac Fibroblast Differentiation
3.3.2. The Effects of Glibenclamide and 5-Hdroxydecanoate on Cardiac Fibroblast Differentiation
3.4. Effects of Ischemia Reperfusion Injury and Ischemic Preconditioning on Fibroblast Differentiation into Immature Vs. Fully Mature Myofibroblasts
4. Discussion
4.1. Characterization of the Ischemic Conditions
4.2. Ischemia Reperfusion Injury and Ischemic Preconditioning in Cardiac Fibroblasts
4.3. The Role of Adenosine Triphosphate-Sensitive Potassium Current in Preventing Fibroblast to Myofibroblast Differentiation
Involvement of Mitochondrial Vs. Sarcolemmal Adenosine Triphosphate-Sensitive Potassium Channels in Ischemic Preconditioning in Fibroblasts
4.4. Immature vs. Fully Mature Myofibroblasts
4.5. Important Considerations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piper, H.M.; Meuter, K.; Schäfer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, 644–648. [Google Scholar] [CrossRef]
- Heyndrickx, G.R. Early reperfusion phenomena. Semin. Cardiothorac. Vasc. Anesth. 2006, 10, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Park, J.L.; Lucchesi, B.R. Mechanisms of myocardial reperfusion injury. Ann. Thorac. Surg. 1999, 68, 1905–1912. [Google Scholar] [CrossRef]
- Murry, C.; Jennings, R.; Reimer, K. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.J.; Peart, J.N. KATP channels and myocardial preconditioning: An update. Am. J. Physiol. Heart Circ. Physiol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, H.; O’Rourke, B. Mitochondrial KATP channels in cell survival and death. J. Mol. Cell Cardiol. 2005, 39, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Sun, X.; Schindler, P.A. The mitochondrial KATP channel as a receptor for potassium channel openers. J. Biol. Chem. 1996, 271, 8796–8799. [Google Scholar] [CrossRef]
- Holmuhamedov, E.L.; Wang, L.; Terzic, A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 1999, 519, 347–360. [Google Scholar] [CrossRef]
- Miyamae, M.; Camacho, S.A.; Weiner, M.W.; Figueredo, V.M. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 1996. [Google Scholar] [CrossRef] [PubMed]
- Negroni, J.A.; Lascano, E.C.; del Valle, H.F. Glibenclamide action on myocardial function and arrhythmia incidence in the healthy and diabetic heart. Cardiovasc. Hematol. Agents. Med. Chem. 2007, 5, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, R.D.; Lodwick, D.; Hudman, D.; Davies, N.W.; Norman, R.I.; Standen, N.B. SUR2A C-terminal fragments reduce KATP currents and ischaemic tolerance of rat cardiac myocytes. J. Physiol 2004, 557, 785–794. [Google Scholar] [CrossRef]
- Grove, D.; Zak, R.; Nair, K.G.; Aschenbrenner, V. Biochemical correlates of cardiac hypertrophy: IV. Observations on the cellular organisation of growth during myocardial hypertrophy in the rat. Circ. Res. 1969, 25, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef]
- Brown, R.D.; Amber, R.C.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Theraputic target in myocardial remodelling and failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Weber, K.T. Infarct scar: A dynamic tissue. Cardiovasc Res. 2000, 46, 250–256. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Diez, J. Fibrosis: A living tissue and the infarcted heart. J. Am. Coll. Cardiol. 2008, 52, 2029–2031. [Google Scholar] [CrossRef]
- Yue, L.; Xie, J.; Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovas. Res. 2011, 89, 744–753. [Google Scholar] [CrossRef]
- Chilton, L.; Ohya, S.; Freed, D.; George, E.; Drobic, V.; Shibukawa, Y.; MacCannell, Y.; Imaizumi, R.B.; Clark, I.; Dixon, M.; et al. K+ currents regulate the resting membrane potential, poliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am. J. Physiol. Heart Circ. Physiol 2005. [Google Scholar] [CrossRef]
- Vander Heide, R.S.; Rim, D.; Hohl, C.M.; Ganote, C.E. An in vitro model of myocardial ischemia utilizing isolated adult rat myocytes. J. Mol. Cell. Cardiol. 1990, 22, 165–181. [Google Scholar] [CrossRef]
- Polewicz, D.; Cadete, V.J.; Doroszko, A.; Hunter, B.E.; Sawicka, J.; Szczesna-Cordary, D.; Light, P.E.; Sawicki, G. Ischemia induced peroxynitrite dependent modifications of cardiomyocyte MLC1 increases its degradation by MMP-2 leading to contractile dysfunction. J. Cell Mol. Med. 2011, 15, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Wang, B.; Jones, S.C.; Jassal, D.S.; Dixon, I.M.C. Interaction between angiotension II and Smad proteins in fibroblasts in failing heart and in vitro. Am. J. Physiol 2000. [Google Scholar] [CrossRef]
- Freed, D.H.; Chilton, L.; Li, Y.; Dangerfield, A.L.; Raizman, J.E.; Rattan, S.G.; Visen, N.; Hryshko, L.V.; Dixon, I.M.C. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am. J. Physiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sato, T.; O’Rourke, B.; Marban, E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation 1998, 97, 2463–2469. [Google Scholar] [CrossRef]
- Saltman, A.E.; Krukenkamp, I.B.; Gaudette, G.R.; Horimoto, H.; Levitsky, S. Pharmacological preconditioning with the adenosine triphosphate-sensitive potassium channel opener pinacidil. Ann. Thorac. Surg. 2000, 70, 595–601. [Google Scholar] [CrossRef]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef]
- Benamer, N.; Fares, N.; Bois, P.; Faivre, J.F. Electrophysiological and functional effects of sphingosine-1-phosphate in mouse ventricular fibroblasts. Biochem. Biophys. Res. Comm. 2011, 408, 6–11. [Google Scholar] [CrossRef]
- Benamer, N.; Moha Ou Maati, H.; Demolombe, S.; Cantereau, A.; Delwail, A.; Bois, P.; Bescond, J.; Faivre, J.F. Molecular and functional characterization of a new potassium conductance in mouse ventricular fibroblasts. J. Mol. Cell Cardiol. 2009, 46, 508–517. [Google Scholar] [CrossRef]
- Benamer, N.; Vasquez, C.; Mahoney, V.M.; Steinhardt, M.J.; Coetzee, W.A.; Morley, G.E. Fibroblast KATP currents modulate myocyte electrophysiology in infarcted hearts. Am. J. Physiol. 2013. [Google Scholar] [CrossRef]
- Fryer, R.M.; Hsu, A.K.; Gross, G.J. Mitochondrial K(ATP) channel opening is important during index ischemia and following myocardial reperfusion in ischemic preconditioned rat hearts. J. Mol. Cell Cardiol 2001, 33, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotech. 2003, 14, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.W.R.; Mifflin, R.C.; Valenich, J.D.; Crowe, S.E.; Saada, J.L.; West, A.B. Myofibroblasts. I. Paracrine cells important in health and disease. Am. J. Physiol. 1999. [Google Scholar] [CrossRef] [PubMed]
- Khuri, S.; Flaherty, J.; O’Riordan, J.; Pitt, B.; Brawley, R.; Donahoo, J.; Gott, V. Changes in intramyocardial ST segment voltage and gas tensions with regional myocardial ischemia in the dog. Circ. Res. 1975, 37, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Cobbe, S.M.; Poole-Wilson, P.A. The time of onset and severity of acidosis in myocardial ischemia. J. Mol. Cell Cardiol. 1980, 12, 745–760. [Google Scholar] [CrossRef]
- Hoffman, J.W., Jr.; Gilbert, T.B.; Poston, R.S.; Silldorff, E.P. Myocardial reperfusion injury: Etiology, mechanisms, and therapies. J. Extra Corpor. Technol. 2004, 36, 391–411. [Google Scholar] [PubMed]
- Vivar, R.; Humeres, C.; Varela, M.; Ayala, P.; Guzmán, N.; Olmedo, I.; Catalán, M.; Boza, P.; Muñoz, C.; Díaz Araya, G. Cardiac fibroblast death by ischemia/reperfusion is partially inhibited by IGF-1 thorugh both PI3K/Akt and MEK-ERK pathways. Exp. Mol. Pathol. 2012, 93, 1–7. [Google Scholar] [CrossRef]
- Zhou, Y.; Richards, A.M.; Wang, P. Characterization and standardization of cultured cardiac fibroblasts for ex vivo models of heart fibrosis and heart ischemia. Tissue Eng. Part. C Methods 2017, 23, 422–433. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, P.; Liu, Q.; Wang, Y.; Zhang, L.; Wu, R.; Chen, J.; Yu, H.; Zhu, W.; Hu, X.; et al. Hepatoma-Derived Growth Factor Secreted from Mesenchymal Stem Cells Reduces Myocardial Ischemia-Reperfusion Injury. Stem Cells Int. 2017. [Google Scholar] [CrossRef]
- Lefort, C.; Benoist, L.; Chadet, S.; Piollet, M.; Heraud, A.; Bebuty, D.; Baron, C.; Ivanes, F.; Angouvant, D. Stimulation of P2Y11 receptor modulates cardiac fibroblasts secretome toward immunomodulatory and protective roles after hypoxia/ reoxygenation injury. J. Mol. Cell. Cardiol. 2018, 121, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, K.; Naganuma, W.; Oqawa, K.; Yaoita, H.; Mizuno, S.; Nakamura, T.; Maruyama, Y. Attenuation of ischemic myocardial injury and dysfunction by fibroblast-derived factor(s). Fukushima, J. Med. Sci. 2010, 56, 1–16. [Google Scholar] [CrossRef]
- Woodall, M.C.; Woodall, B.P.; Gao, E.; Yuan, A.; Koch, W.J. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ. Res. 2016, 119, 1116–1127. [Google Scholar] [CrossRef]
- Yao, Z.; Gross, G. Effects of the KATP channel opener bimakalim on coronary blood flow, monophasic action potential duration, and infarct size in dogs. Circulation 1994, 89, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Chatelier, A.; Mercier, A.; Tremblier, B.; Theriault, O.; Moubarak, M.; Benamer, N.; Corbe, P.; Bois, P.; Chahine, M.; Faivre, J.F. A distinct de novo expression of Nav1.5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J. Physiol. 2012, 590, 4307–4319. [Google Scholar] [CrossRef]
- Chilton, L.; Giles, W.R.; Smith, G.L. Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J. Physiol. 2007, 583, 225–236. [Google Scholar] [CrossRef]
- Rook, M.B.; van Ginneken, A.C.G.; de Jonge, B.; El Aoumari, A.; Gros, D.; Jongsma, H.J. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am. J. Physiol. 1992. [Google Scholar] [CrossRef]
- Raizman, J.E.; Komljenovic, J.; Chang, R.; Deng, C.; Bedosky, K.M.; Rattan, S.G.; Cunnington, R.H.; Freed, D.H.; Dixon, I.M.C. The participation of the Na+-Ca2+ exchanger in primary cardiac myofibroblast migration, contraction, and proliferation. J. Cell. Physiol. 2007, 231, 540–551. [Google Scholar] [CrossRef]
- Calderone, A.; Thalk, C.M.; Takahashi, N.; Change, D.L.F.; Colucci, W.S. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Invest. 1998, 101, 812–818. [Google Scholar] [CrossRef]
- Shivakumar, K.; Kumaran, C. L-type calcium channel blockers and EGTA ehance superoxide production in cardiac fibroblasts. J. Mol. Cell Cardiol 2001, 33, 373–377. [Google Scholar] [CrossRef]
- Dawson, K.; Wu, C.T.; Qi, X.Y.; Nattel, S. Congestive heart failure effects on atrial fibroblast phenotype: Differences between freshly-isolated and cultured cells. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.B.; Zhang, J. Neonatal rat cardiac fibroblasts express three types of voltage-gated K+ channels: Regulation of a transient outward current by protein kinase C. Am. J. Physiol. Heart Circ. Physiol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Kamkin, A.; Kirischuk, S.; Kiseleva, I. Single mechano-gated channels activated by mechanical deformation of acutely isolated cardiac fibroblasts from rats. Acta. Physiol. 2010, 199, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Kamkin, A.; Kiseleva, I.; Isenberg, G. Activation and inactivation of a nonselective cation conductance by local mechanical deformation in acutely isolated cardiac fibroblasts. Cardiovasc. Res. 2003, 57, 793–803. [Google Scholar] [CrossRef]
- Li, G.R.; Sun, H.Y.; Chen, J.B.; Zhou, Y.; Tse, H.F.; Lau, C.P. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Shim, W.; Wei, H.; Lim, S.Y.; Liew, R.; Lim, T.S.; Ong, B.H.; Chua, Y.L.; Wong, P. Hydrogen sulphide suppresses human atrial fibroblast proliferation and transformation to myofibroblasts. J. Cell Mol. Med. 2013, 17, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Shibukawa, Y.; Chilton, E.L.; MacCannell, K.A.; Clark, R.B.; Giles, W.R. K+ currents activated by depolarization in cardiac fibroblasts. Biophysics, J. 2005, 88, 3924–3935. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, C.; Mohandas, P.; Louie, K.L.; Benamer, N.; Bapat, A.C.; Morley, G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010, 107, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Wang, Y.; Zhao, L.M.; Li, G.R.; Deng, X.L. Angiotensin II upregulates KCa3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts. Biochem. Pharmacol. 2013, 85, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, N.; Pepper, M.S.; Modarressi, A.; Alfo, K.; Schlaudraff, K.; Montandon, D.; Gabbiani, G.; Bochaton-Piallat, M.L.; Pittet, B. Persistent ischemia impairs myofibroblast development in wound granulation tissue: A new model of delayed wound healing. Wound Rep. Reg 2007, 15, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Steinbrech, D.S.; Longaker, M.T.; Mehrara, B.J.; Saadeh, P.B.; Chin, G.S.; Gerrets, M.A.; Chau, D.C.; Rowe, M.N.; Gittes, G.K. Fibroblast response to hypoxia: The relationship between angiogenesis and matrix regulation. J. Surg Res. 1999, 84, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Ishikawa, O. Hypoxic conditions decrease the mRNA expression of proα1(I) and (III) collagens and increase matrix metalloproteinases-1 of dermal fibroblasts in three-dimensional cultures. J. Dermatol Sci 2000, 24, 99–104. [Google Scholar] [CrossRef]
- Li, X.; Misik, A.J.; Solaro, R.J.; Lowen, A.; Lieigel, L. Thyroid hormone receptor alpha 1 regulates expression of the Na+/H+ exchanger (NHE1). J. Biol. Chem. 2002, 277, 28656–28662. [Google Scholar] [CrossRef] [PubMed]
- D’hahan, N.; Moreau, C.; Prost, A.L.; Jacquet, H.; Alekseev, A.E.; Terzic, A.; Vivaudou, M. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc. Natl. Acad. Sci. USA 1999, 96, 12162–12167. [Google Scholar] [CrossRef] [PubMed]
- Kersten, J.R.; Schmeling, T.J.; Pagel, P.S.; Gross, G.J.; Warltier, D.C. Isoflurane mimics ischemic preconditioning via activation of KATP channels: Reduction of myocardial infarct size with an acute memory phase. Anesthesiology 1997, 87, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Redel, A.; Stumpner, J.; Tischer-Zeitz, T.; Lange, M.; Smul, T.M.; Lotz, C.; Roewer, N.; Kehl, F. Comparison of isoflurane-, sevoflurane-, and desflurane-induced pre- and postconditioning against myocardial infarction in mice in vivo. Exp. Biol. Med. 2009, 234, 1186–1191. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pertiwi, K.R.; Hillman, R.M.; Scott, C.A.; Chilton, E.L. Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of KATP Channels. J. Cardiovasc. Dev. Dis. 2019, 6, 22. https://doi.org/10.3390/jcdd6020022
Pertiwi KR, Hillman RM, Scott CA, Chilton EL. Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of KATP Channels. Journal of Cardiovascular Development and Disease. 2019; 6(2):22. https://doi.org/10.3390/jcdd6020022
Chicago/Turabian StylePertiwi, Kartika R., Rachael M. Hillman, Coralie A. Scott, and Emily Lisa Chilton. 2019. "Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of KATP Channels" Journal of Cardiovascular Development and Disease 6, no. 2: 22. https://doi.org/10.3390/jcdd6020022
APA StylePertiwi, K. R., Hillman, R. M., Scott, C. A., & Chilton, E. L. (2019). Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of KATP Channels. Journal of Cardiovascular Development and Disease, 6(2), 22. https://doi.org/10.3390/jcdd6020022