Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling


Abstract
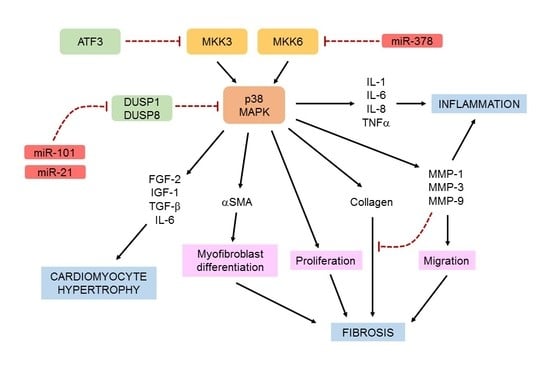
{kind=link}
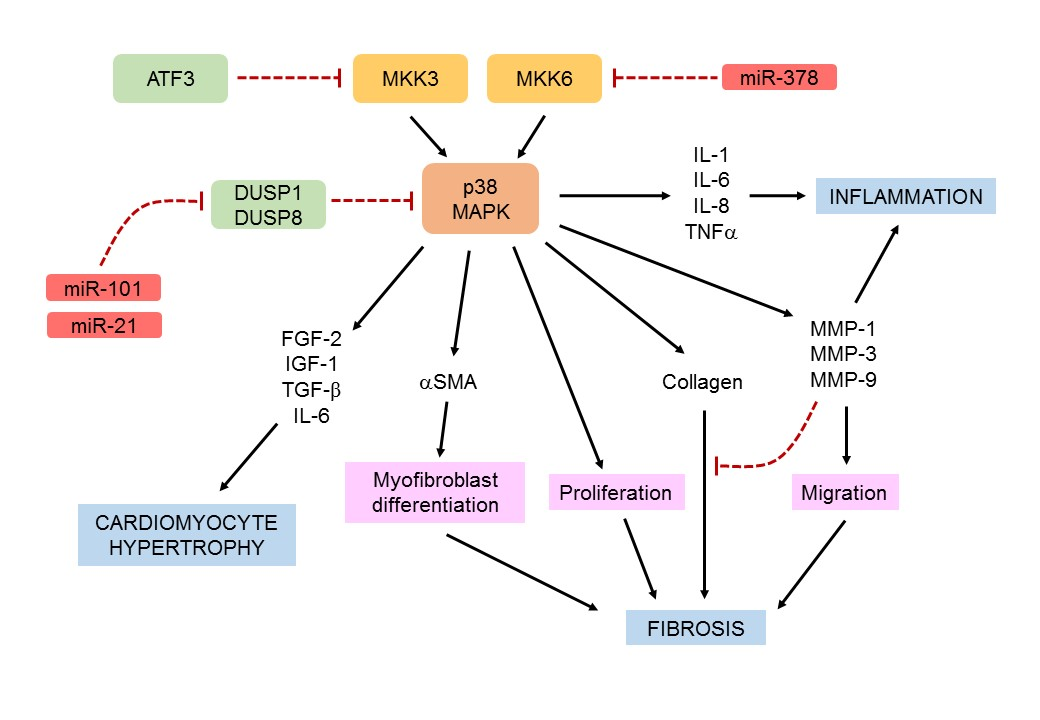
{kind=link}
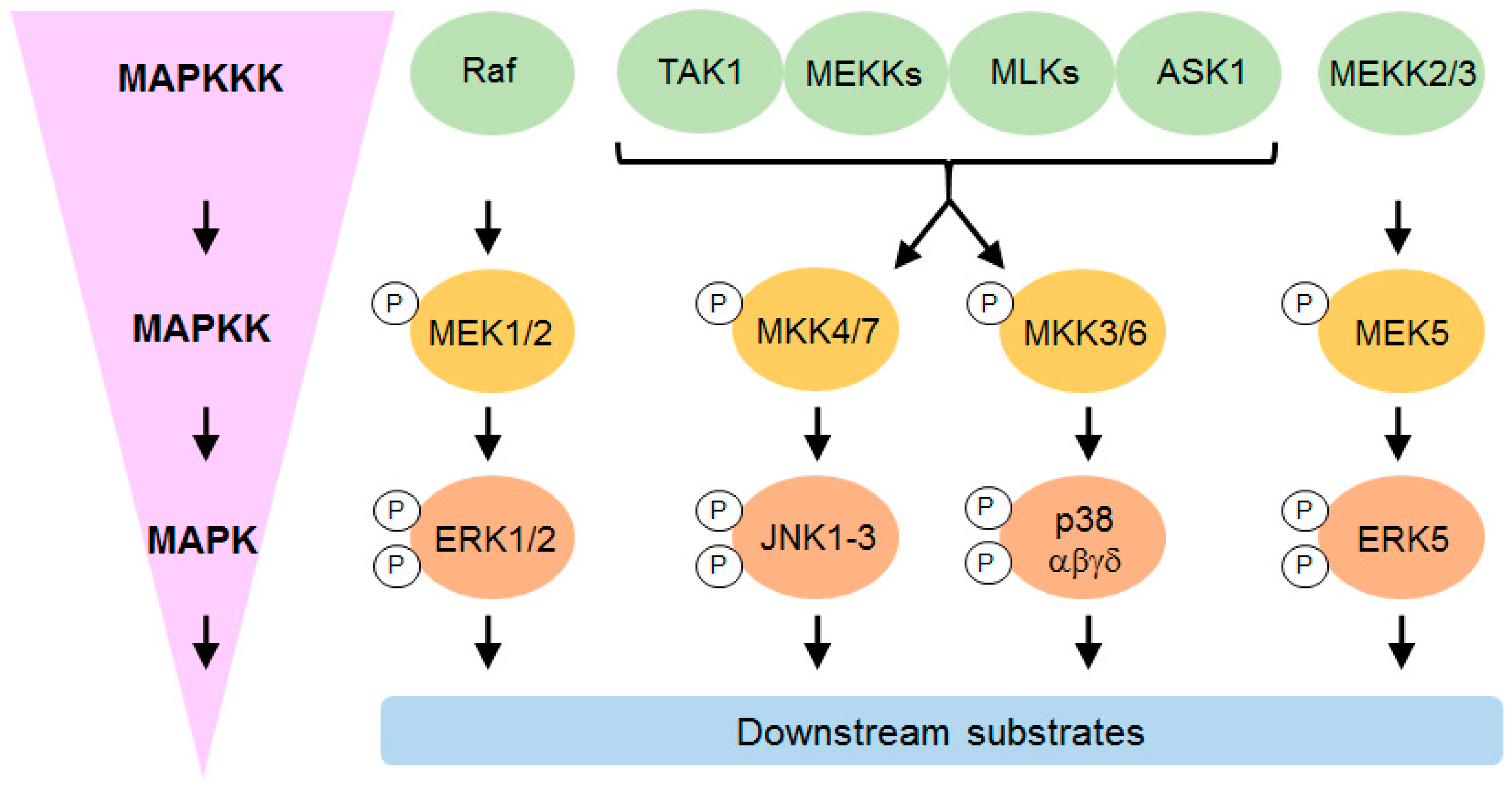
{kind=link}
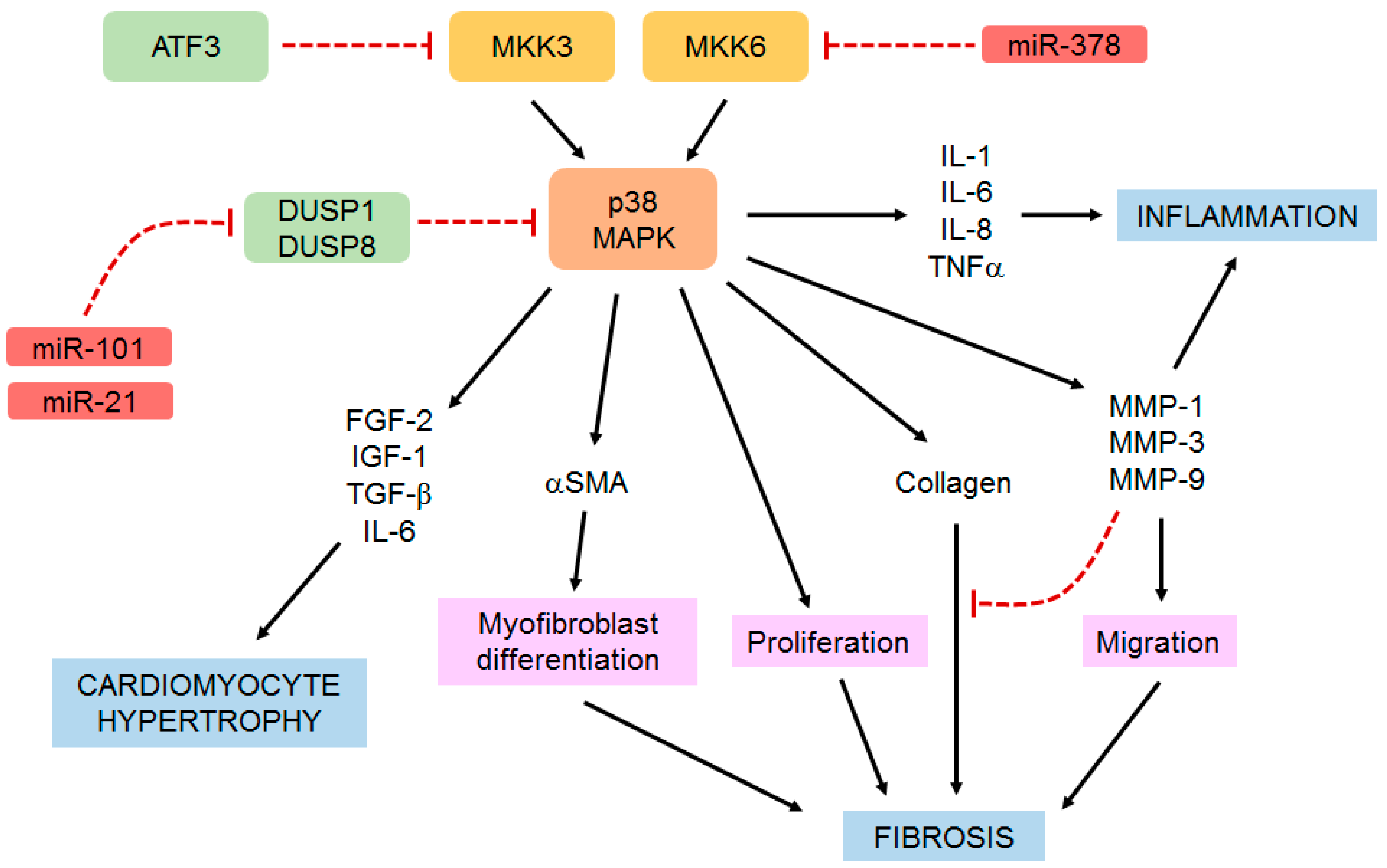{kind=link}
1. Introduction
2. Cardiac Fibroblasts and Myocardial Remodeling
3. p38 MAP Kinase
3.1. MAP Kinase Signaling Cascades
3.2. p38 MAP Kinase Signaling
3.2.1. p38 Subtypes and Their Activation
3.2.2. Modulation of Gene and Protein Expression
3.2.3. MAPK-Activated Protein Kinases
3.2.4. MAPK Phosphatases
4. p38 MAP Kinase and Regulation of Cardiac Remodeling
4.1. In Vitro and In Vivo Studies
4.2. Clinical Studies
5. p38 MAP Kinase and Regulation of Cardiac Fibroblast Function
5.1. In Vitro Studies
5.1.1. Proinflammatory Cytokines
5.1.2. Hypertrophic Factors
5.1.3. Matrix Metalloproteinases
5.1.4. α-Smooth Muscle Actin
5.1.5. Proliferation, Migration and Myofibroblast Differentiation
5.2. In Vivo Studies
5.2.1. Fibroblast-Specific Targeting of p38α
5.2.2. Fibroblast-Specific Targeting of Other Components of the p38 Pathway
5.2.3. MicroRNAs
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Dostal, D.; Glaser, S.; Baudino, T.A. Cardiac fibroblast physiology and pathology. Compr. Physiol. 2015, 5, 887–909. [Google Scholar] [PubMed]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway-A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Wang, Y. p38 MAP kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Van den Borne, S.W.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 5. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J.; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac fibroblast activation post-myocardial infarction: Current knowledge gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Porter, K.E. Regulation of myocardial matrix metalloproteinase expression and activity by cardiac fibroblasts. IUBMB Life 2012, 64, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- MacKenna, D.; Summerour, S.R.; Villarreal, F.J. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc. Res. 2000, 46, 257–263. [Google Scholar] [CrossRef]
- Fujiu, K.; Nagai, R. Fibroblast-mediated pathways in cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 70, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kamo, T.; Akazawa, H.; Komuro, I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ. Res. 2015, 117, 89–98. [Google Scholar] [CrossRef]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell. Mol. Life Sci. 2008, 65, 3525–3544. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Lee, J.D. Role of the BMK1/ERK5 signaling pathway: Lessons from knockout mice. J. Mol. Med. 2004, 82, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Mitogen-activated protein kinases in heart development and diseases. Circulation 2007, 116, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A. Therapeutic regulation of cardiac fibroblast function: Targeting stress-activated protein kinase pathways. Future Cardiol. 2011, 7, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Bassi, R.; Gorog, D.A.; Saurin, A.T.; Jiang, J.; Heads, R.J.; Martin, J.L.; Davis, R.J.; Flavell, R.A.; Marber, M.S. Diverse mechanisms of myocardial p38 mitogen-activated protein kinase activation: Evidence for MKK-independent activation by a TAB1-associated mechanism contributing to injury during myocardial ischemia. Circ. Res. 2003, 93, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Kang, Y.J.; Han, J.; Herschman, H.R.; Stefani, E.; Wang, Y. TAB-1 modulates intracellular localization of p38 MAP kinase and downstream signaling. J. Biol. Chem. 2006, 281, 6087–6095. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shang, E.; Dong, Q.; Li, Y.; Zhang, J.; Xu, S.; Zhao, Z.; Shao, W.; Lv, C.; Zheng, Y.; et al. Small molecules capable of activating DNA methylation-repressed genes targeted by the p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 2018, 293, 7423–7436. [Google Scholar] [CrossRef]
- Vermeulen, L.; Vanden Berghe, W.; Beck, I.M.; De, B.K.; Haegeman, G. The versatile role of MSKs in transcriptional regulation. Trends Biochem. Sci. 2009, 34, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Cruzalegui, F.H.; Treisman, R. The p38 and ERK MAP kinase pathways cooperate to activate Ternary Complex Factors and c-fos transcription in response to UV light. EMBO J. 1996, 15, 6552–6563. [Google Scholar] [CrossRef]
- Dean, J.L.; Sully, G.; Clark, A.R.; Saklatvala, J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell. Signal. 2004, 16, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.D.; Ammit, A.J.; Clark, A.R. MAPK p38 regulates inflammatory gene expression via tristetraprolin: Doing good by stealth. Int. J. Biochem. Cell Biol. 2018, 94, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Platanias, L.C. Mnk kinase pathway: Cellular functions and biological outcomes. World J. Biol. Chem. 2014, 5, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Kotlyarov, A.; Gaestel, M. MK2 and MK3-a pair of isoenzymes? Front. Biosci. 2008, 13, 5511–5521. [Google Scholar] [CrossRef] [PubMed]
- Gaestel, M. MAPKAP kinases-MKs-two’s company, three’s a crowd. Nat. Rev. Mol. Cell Biol. 2006, 7, 120–130. [Google Scholar] [CrossRef]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, A.; Moens, U. Mitogen-activated protein kinase p38 and MK2, MK3 and MK5: Menage a trois or menage a quatre? Cell. Signal. 2010, 22, 1185–1192. [Google Scholar] [CrossRef]
- Aberg, E.; Perander, M.; Johansen, B.; Julien, C.; Meloche, S.; Keyse, S.M.; Seternes, O.M. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 2006, 281, 35499–35510. [Google Scholar] [CrossRef]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef]
- Liu, R.; Molkentin, J.D. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J. Mol. Cell. Cardiol. 2016, 101, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Bassi, R.; Marber, M.S. p38 MAPK in cardioprotection-are we there yet? Br. J. Pharmacol. 2015, 172, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Arabacilar, P.; Marber, M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front. Pharmacol. 2015, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Bhandary, B.; Osinska, H.; James, J.; Xu, N.; Shay-Winkler, K.; Gulick, J.; Willis, M.S.; Lander, C.; Robbins, J. MMI-0100 inhibits cardiac fibrosis in a mouse model overexpressing cardiac myosin binding protein C. J. Am. Heart Assoc. 2017, 6, e006590. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yates, C.C.; Lockyer, P.; Xie, L.; Bevilacqua, A.; He, J.; Lander, C.; Patterson, C.; Willis, M. MMI-0100 inhibits cardiac fibrosis in myocardial infarction by direct actions on cardiomyocytes and fibroblasts via MK2 inhibition. J. Mol. Cell. Cardiol. 2014, 77, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Cooley, B.C.; Quintana, M.T.; Lander, C.; Willis, M.S. Nebulized delivery of the MAPKAP kinase 2 peptide inhibitor MMI-0100 protects against ischemia-induced systolic dysfunction. Int. J. Pept. Res. Ther. 2016, 22, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Clerk, A.; Sugden, P.H. Inflame my heart (by p38-MAPK). Circ. Res. 2006, 99, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, E.; Kaikkonen, L.; Tokola, H.; Pikkarainen, S.; Aro, J.; Pennanen, H.; Karvonen, T.; Rysa, J.; Kerkela, R.; Ruskoaho, H. Distinct regulation of B-type natriuretic peptide transcription by p38 MAPK isoforms. Mol. Cell. Endocrinol. 2011, 338, 18–27. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, S.; Sah, V.P.; Ross, J., Jr.; Brown, J.H.; Han, J.; Chien, K.R. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 1998, 273, 2161–2168. [Google Scholar] [CrossRef]
- Braz, J.C.; Bueno, O.F.; Liang, Q.; Wilkins, B.J.; Dai, Y.S.; Parsons, S.; Braunwart, J.; Glascock, B.J.; Klevitsky, R.; Kimball, T.F.; et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Investig. 2003, 111, 1475–1486. [Google Scholar] [CrossRef]
- Nishida, K.; Yamaguchi, O.; Hirotani, S.; Hikoso, S.; Higuchi, Y.; Watanabe, T.; Takeda, T.; Osuka, S.; Morita, T.; Kondoh, G.; et al. p38a mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 2004, 24, 10611–10620. [Google Scholar] [CrossRef]
- Sari, F.R.; Widyantoro, B.; Thandavarayan, R.A.; Harima, M.; Lakshmanan, A.P.; Zhang, S.; Muslin, A.J.; Suzuki, K.; Kodama, M.; Watanabe, K. Attenuation of CHOP-mediated myocardial apoptosis in pressure-overloaded dominant negative p38a mitogen-activated protein kinase mice. Cell. Physiol. Biochem. 2011, 27, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinheimer, C.; Courtois, M.; Kovacs, A.; Zhang, C.E.; Cheng, A.M.; Wang, Y.; Muslin, A.J. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Investig. 2003, 111, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Teran, B.; Lopez, J.A.; Rodriguez, E.; Leiva, L.; Martinez-Martinez, S.; Bernal, J.A.; Jimenez-Borreguero, L.J.; Redondo, J.M.; Vazquez, J.; Sabio, G. p38g and d promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016, 7, 10477. [Google Scholar] [CrossRef] [PubMed]
- Auger-Messier, M.; Accornero, F.; Goonasekera, S.A.; Bueno, O.F.; Lorenz, J.N.; van Berlo, J.H.; Willette, R.N.; Molkentin, J.D. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 2013, 112, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Newby, L.K.; Marber, M.S.; Melloni, C.; Sarov-Blat, L.; Aberle, L.H.; Aylward, P.E.; Cai, G.; de Winter, R.J.; Hamm, C.W.; Heitner, J.F.; et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial. Lancet 2014, 384, 1187–1195. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.; Gutierrez, J.A.; et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: A randomized clinical trial. JAMA 2016, 315, 1591–1599. [Google Scholar] [CrossRef]
- Sinfield, J.K.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E.; Turner, N.A. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2013, 430, 419–424. [Google Scholar] [CrossRef]
- Lemke, L.E.; Bloem, L.J.; Fouts, R.; Esterman, M.; Sandusky, G.; Vlahos, C.J. Decreased p38 MAPK activity in end-stage failing human myocardium: p38 MAPK a is the predominant isoform expressed in human heart. J. Mol. Cell. Cardiol. 2001, 33, 1527–1540. [Google Scholar] [CrossRef]
- Dingar, D.; Merlen, C.; Grandy, S.; Gillis, M.A.; Villeneuve, L.R.; Mamarbachi, A.M.; Fiset, C.; Allen, B.G. Effect of pressure overload-induced hypertrophy on the expression and localization of p38 MAP kinase isoforms in the mouse heart. Cell. Signal. 2010, 22, 1634–1644. [Google Scholar] [CrossRef]
- Turner, N.A. Effects of interleukin-1 on cardiac fibroblast function: Relevance to post-myocardial infarction remodelling. Vascul. Pharmacol. 2014, 60, 1–7. [Google Scholar] [CrossRef] [PubMed]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol. 2004, 44, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhong, M.; Shang, Y.; Lin, H.; Deng, J.; Jiang, H.; Lu, H.; Zhang, Y.; Zhang, W. Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell. Mol. Life Sci. 2008, 65, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G.; et al. p38 MAPK inhibition improves heart function in pressure-loaded right ventricular hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.W.; Kvakan, H.; Kaun, C.; Zorn, G.; Speidl, W.S.; Pfaffenberger, S.; Maurer, G.; Huber, K.; Wojta, J. The gp130 ligand oncostatin M regulates tissue inhibitor of metalloproteinases-1 through ERK1/2 and p38 in human adult cardiac myocytes and in human adult cardiac fibroblasts: A possible role for the gp130/gp130 ligand system in the modulation of extracellular matrix degradation in the human heart. J. Mol. Cell. Cardiol. 2005, 39, 545–551. [Google Scholar] [PubMed]
- Turner, N.A.; Das, A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Interleukin-1a stimulates pro-inflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1117–H1127. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Mechanism of TNFa-induced IL-1a, IL-1b and IL-6 expression in human cardiac fibroblasts: Effects of statins and thiazolidinediones. Cardiovasc. Res. 2007, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Human cardiac fibroblasts express ICAM-1, E-selectin and CXC chemokines in response to proinflammatory cytokine stimulation. Int. J. Biochem. Cell Biol. 2011, 43, 1450–1458. [Google Scholar] [CrossRef]
- Tang, W.; Wei, Y.; Le, K.; Li, Z.; Bao, Y.; Gao, J.; Zhang, F.; Cheng, S.; Liu, P. Mitogen-activated protein kinases ERK 1/2- and p38-GATA4 pathways mediate the Ang II-induced activation of FGF2 gene in neonatal rat cardiomyocytes. Biochem. Pharmacol. 2011, 81, 518–525. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, T.; Zhang, K.; Liu, Y.; Huang, F.; Zhu, X.; Liu, Y.; Wang, M.H.; Tang, W.; Wang, J.; et al. Deletion of soluble epoxide hydrolase attenuates cardiac hypertrophy via down-regulation of cardiac fibroblasts-derived fibroblast growth factor-2. Crit. Care Med. 2014, 42, e345–e354. [Google Scholar] [CrossRef]
- Jiao, S.; Ren, H.; Li, Y.; Zhou, J.; Duan, C.; Lu, L. Differential regulation of IGF-I and IGF-II gene expression in skeletal muscle cells. Mol. Cell. Biochem. 2013, 373, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Q.; Freire-de-Lima, C.G.; Schiemann, W.P.; Bratton, D.L.; Vandivier, R.W.; Henson, P.M. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J. Immunol. 2008, 181, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cheng, G.; Jin, R.; Afzal, M.R.; Samanta, A.; Xuan, Y.T.; Girgis, M.; Elias, H.K.; Zhu, Y.; Davani, A.; et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ. Res. 2016, 118, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Ancey, C.; Menet, E.; Corbi, P.; Fredj, S.; Garcia, M.; Rucker-Martin, C.; Bescond, J.; Morel, F.; Wijdenes, J.; Lecron, J.C.; et al. Human cardiomyocyte hypertrophy induced in vitro by gp130 stimulation. Cardiovasc. Res. 2003, 59, 78–85. [Google Scholar] [CrossRef]
- Tenhunen, O.; Rysa, J.; Ilves, M.; Soini, Y.; Ruskoaho, H.; Leskinen, H. Identification of cell cycle regulatory and inflammatory genes as predominant targets of p38 mitogen-activated protein kinase in the heart. Circ. Res. 2006, 99, 485–493. [Google Scholar] [CrossRef]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; et al. ERK and p38 MAPK, but not NF-kB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, M.; Kirkwood, K.L. p38alpha stabilizes interleukin-6 mRNA via multiple AU-rich elements. J. Biol. Chem. 2008, 283, 1778–1785. [Google Scholar] [CrossRef]
- Van Tubergen, E.A.; Banerjee, R.; Liu, M.; Vander, B.R.; Light, E.; Kuo, S.; Feinberg, S.E.; Willis, A.L.; Wolf, G.; Carey, T.; et al. Inactivation or loss of TTP promotes invasion in head and neck cancer via transcript stabilization and secretion of MMP9, MMP2, and IL-6. Clin. Cancer Res. 2013, 19, 1169–1179. [Google Scholar] [CrossRef]
- Ohkura, S.I.; Usui, S.; Takashima, S.I.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Inagaki, Y.; Sugimoto, N.; Kitano, T.; Takamura, M.; et al. Augmented sphingosine 1 phosphate receptor-1 signaling in cardiac fibroblasts induces cardiac hypertrophy and fibrosis through angiotensin II and interleukin-6. PLoS ONE 2017, 12, e0182329. [Google Scholar] [CrossRef]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38a MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol. Rev. 2007, 87, 1285–1342. [Google Scholar] [CrossRef] [PubMed]
- Fanjul-Fernandez, M.; Folgueras, A.R.; Cabrera, S.; Lopez-Otin, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Lisanti, M.P.; Scherer, P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998, 273, 32111–32120. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.M.; Feldman, M.D.; Mummidi, S.; Valente, A.J.; Steffensen, B.; Vincenti, M.; Barnes, J.L.; Chandrasekar, B. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK-and ERK1/2-dependent C/EBP-b, NF-kB, and AP-1 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3356–H3365. [Google Scholar] [CrossRef] [PubMed]
- Reunanen, N.; Li, S.P.; Ahonen, M.; Foschi, M.; Han, J.; Kahari, V.M. Activation of p38a MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J. Biol. Chem. 2002, 277, 32360–32368. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Modulatory effect of interleukin-1a on expression of structural matrix proteins, MMPs and TIMPs in human cardiac myofibroblasts: Role of p38 MAP kinase. Matrix Biol. 2010, 29, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Jones, G.M.; Laird, R.E.; Hudson, P.; Long, C.S. Cytokines regulate matrix metalloproteinases and migration in cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2007, 362, 200–205. [Google Scholar] [CrossRef]
- Eberhardt, W.; Huwiler, A.; Beck, K.F.; Walpen, S.; Pfeilschifter, J. Amplification of IL-1 beta-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-kappa B and activating protein-1 and involves activation of the mitogen-activated protein kinase pathways. J. Immunol. 2000, 165, 5788–5797. [Google Scholar]
- Simon, C.; Simon, M.; Vucelic, G.; Hicks, M.J.; Plinkert, P.K.; Koitschev, A.; Zenner, H.P. The p38 SAPK pathway regulates the expression of the MMP-9 collagenase via AP-1-dependent promoter activation. Exp. Cell Res. 2001, 271, 344–355. [Google Scholar] [CrossRef]
- Kumar, B.; Koul, S.; Petersen, J.; Khandrika, L.; Hwa, J.S.; Meacham, R.B.; Wilson, S.; Koul, H.K. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010, 70, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Akool, E.-S.; Kleinert, H.; Hamada, F.M.A.; Abdelwahab, M.H.; Förstermann, U.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol. Cell. Biol. 2003, 23, 4901–4916. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Penke, L.R.; Huang, S.K.; White, E.S.; Peters-Golden, M. Prostaglandin E2 inhibits a-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J. Biol. Chem. 2014, 289, 17151–17162. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Menon, M.B.; Schwermann, J.; Arthur, J.S.; Legault, H.; Telliez, J.B.; Kayyali, U.S.; Nebreda, A.R.; Kotlyarov, A.; Gaestel, M. Stress induced gene expression: A direct role for MAPKAP kinases in transcriptional activation of immediate early genes. Nucleic Acids Res. 2011, 39, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, X.; Wei, S.M.; Tang, Y.H.; Zhou, Q.; Huang, C.X. Activin A stimulates the proliferation and differentiation of cardiac fibroblasts via the ERK1/2 and p38-MAPK pathways. Eur. J. Pharmacol. 2016, 789, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J.M. Fibroblast-specific genetic manipulation of p38 MAPK in vivo reveals its central regulatory role in fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Liang, J.; Liu, N.; Liu, X.; Mena, J.M.; Xie, T.; Geng, Y.; Huan, C.; Zhang, Y.; Taghavifar, F.; Huang, G.; et al. Mitogen-activated protein kinase-activated protein kinase 2 inhibition attenuates fibroblast invasion and severe lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 41–48. [Google Scholar] [CrossRef]
- Sousa, A.M.; Liu, T.; Guevara, O.; Stevens, J.; Fanburg, B.L.; Gaestel, M.; Toksoz, D.; Kayyali, U.S. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J. Cell. Biochem. 2007, 100, 1581–1592. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Delafontaine, P.; Chandrasekar, B. Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell. Signal. 2012, 24, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Duan, J.; Pillai, I.C.; Rosa-Garrido, M.; Wu, Y.; Bargiacchi, F.; Lu, Y.; Stanbouly, S.; Huang, J.; Rojas, M.; et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 2014, 514, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Aronow, B.J.; Tallquist, M.D.; Molkentin, J.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Kuwabara, J.T.; Pai, J.T.; Moore, R.E.; Sun, Z.; Tallquist, M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 2018, 114, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3b regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.L.; Fang, M.; Yutzey, K.E. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Zhang, C.; Li, P.; Wu, Y.; Wang, C.; Lau, W.B.; Ma, X.L.; Du, J. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 signaling. Circulation 2017, 135, 2041–2057. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Hao, W.J.; Chen, B.Y.; Chen, J.; Li, G.Q. Cardiac fibroblast-specific activating transcription factor 3 promotes myocardial repair after myocardial infarction. Chin. Med. J. 2018, 131, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, P.; Allen, B.G. MK5: A novel regulator of cardiac fibroblast function? IUBMB Life 2017, 69, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Nawaito, S.A.; Dingar, D.; Sahadevan, P.; Hussein, B.; Sahmi, F.; Shi, Y.; Gillis, M.A.; Gaestel, M.; Tardif, J.C.; Allen, B.G. MK5 haplodeficiency attenuates hypertrophy and preserves diastolic function during remodeling induced by chronic pressure overload in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H46–H58. [Google Scholar] [CrossRef] [PubMed]
- Dingar, D.; Benoit, M.J.; Mamarbachi, A.M.; Villeneuve, L.R.; Gillis, M.A.; Grandy, S.; Gaestel, M.; Fiset, C.; Allen, B.G. Characterization of the expression and regulation of MK5 in the murine ventricular myocardium. Cell. Signal. 2010, 22, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.T.; Bar, C.; Thum, T. Non-coding RNAs as modulators of the cardiac fibroblast phenotype. J. Mol. Cell. Cardiol. 2016, 92, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, W.; Xu, M.; Huang, H.; Wang, J.; Chen, X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can. J. Cardiol. 2014, 30, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 2018, 8, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from bench to clinic as next generation medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. https://doi.org/10.3390/jcdd6030027
Turner NA, Blythe NM. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. Journal of Cardiovascular Development and Disease. 2019; 6(3):27. https://doi.org/10.3390/jcdd6030027
Chicago/Turabian StyleTurner, Neil A., and Nicola M. Blythe. 2019. "Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling" Journal of Cardiovascular Development and Disease 6, no. 3: 27. https://doi.org/10.3390/jcdd6030027
APA StyleTurner, N. A., & Blythe, N. M. (2019). Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. Journal of Cardiovascular Development and Disease, 6(3), 27. https://doi.org/10.3390/jcdd6030027