The Role of Non-Coding RNA in Congenital Heart Diseases



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. microRNAs and Cardiac Congenital Heart Diseases
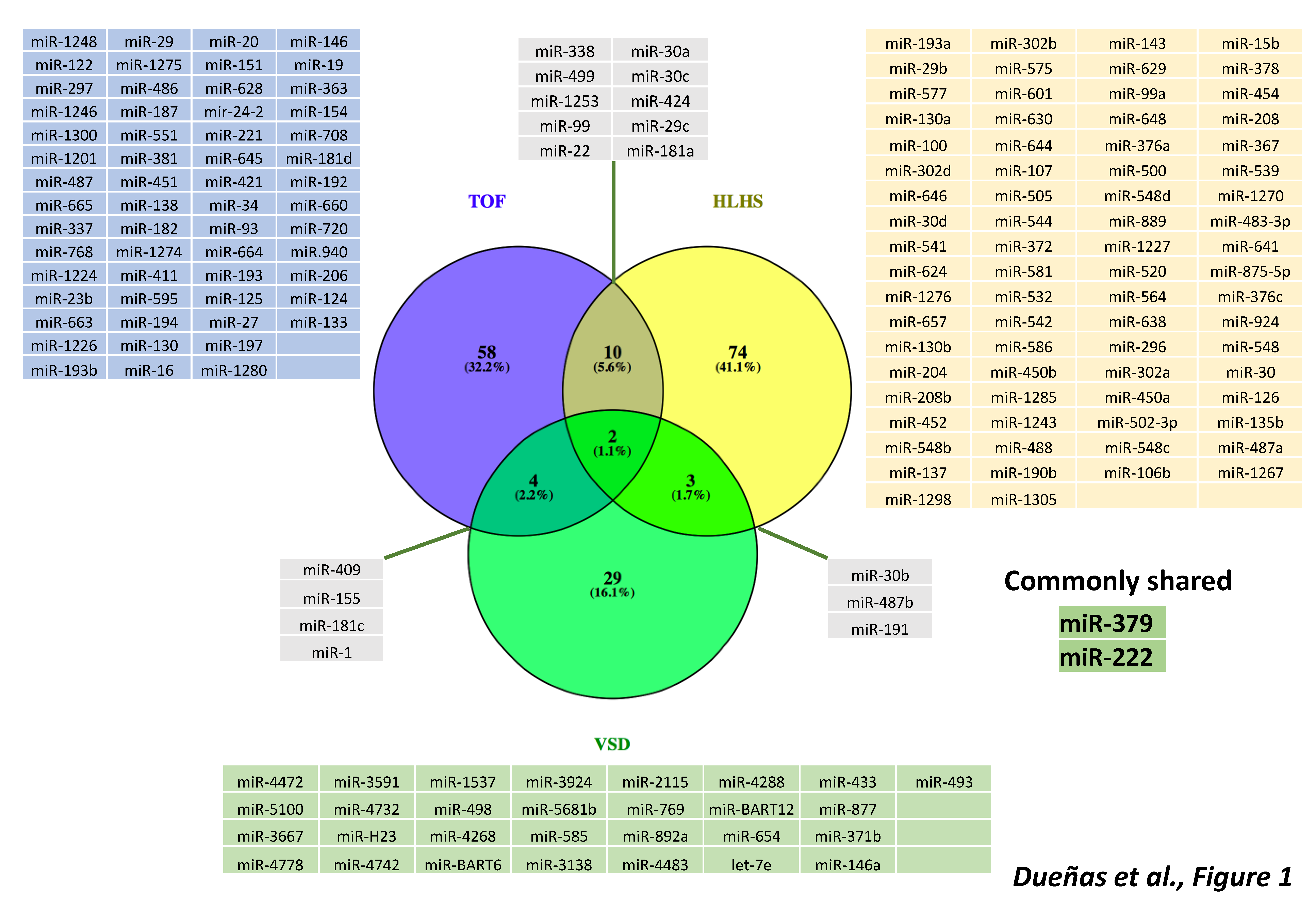
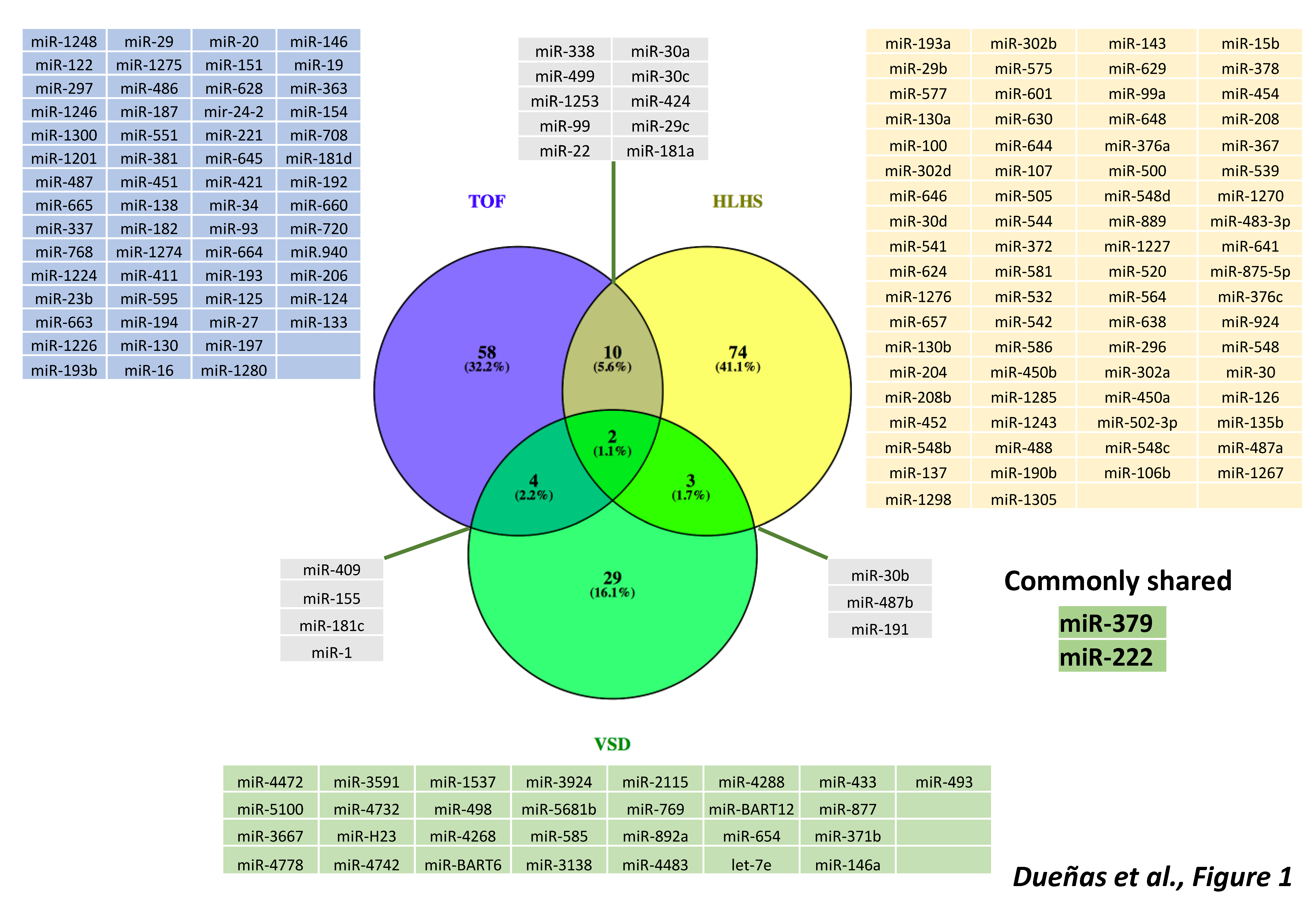
3. microRNAs in Cyanotic Heart Diseases
4. microRNAs in Left-Sided Cardiac Obstructions
5. microRNAs in Septal Defects
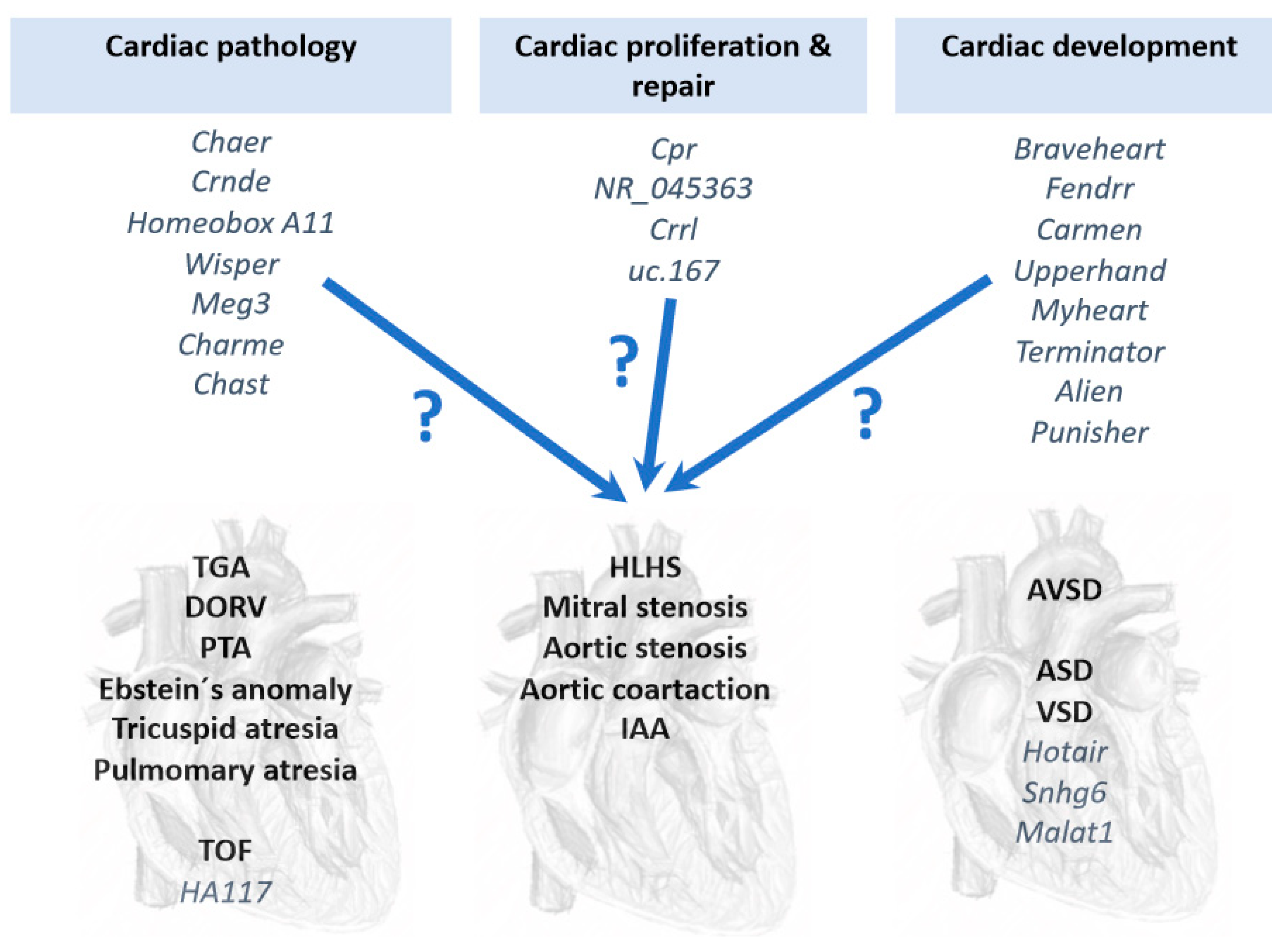
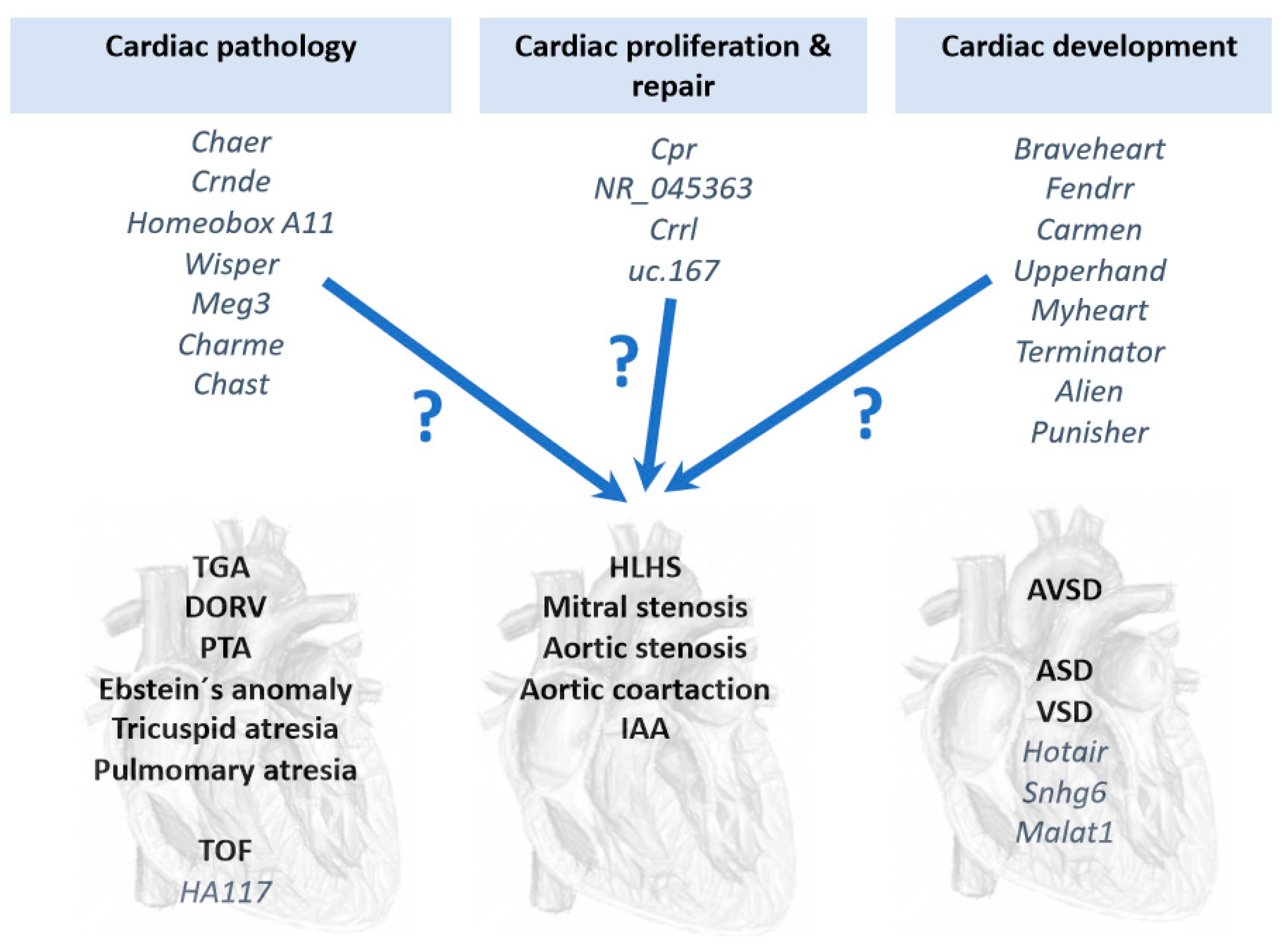
6. Long Non-Coding RNAs (lncRNAs) in Congenital Heart Diseases
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- López-Sánchez, C.; García-Martínez, V. Molecular determinants of cardiac specification. Cardiovasc. Res. 2011, 91, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Christoffels, V.M.; Campione, M. Homeobox transcription factor Pitx2: The rise of an asymmetry gene in cardiogenesis and arrhythmogenesis. Trends Cardiovasc. Med. 2014, 24, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Campione, M.; Franco, D. Current Perspectives in Cardiac Laterality. J. Cardiovasc. Dev. Dis. 2016, 3, 34. [Google Scholar] [CrossRef]
- Dueñas, A.; Aranega, A.E.; Franco, D. More than Just a Simple Cardiac Envelope; Cellular Contributions of the Epicardium. Front. Cell Dev. Biol. 2017, 5, 44. [Google Scholar] [CrossRef]
- Miyagawa-Tomita, S.; Arima, Y.; Kurihara, H. The “Cardiac Neural Crest” Concept Revisited. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; Chapter 30. [Google Scholar]
- Moorman, A.F.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Captur, G.; Syrris, P.; Obianyo, C.; Limongelli, G.; Moon, J.C. Formation and Malformation of Cardiac Trabeculae: Biological Basis, Clinical Significance, and Special Yield of Magnetic Resonance Imaging in Assessment. Can. J. Cardiol. 2015, 31, 1325–1337. [Google Scholar] [CrossRef]
- D’Amato, G.; Luxán, G.; de la Pompa, J.L. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J. 2016, 283, 4223–4237. [Google Scholar] [CrossRef]
- Gelb, B.D. Genetic Discovery for Congenital Heart Defects. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; Chapter 51. [Google Scholar]
- Russell, M.W.; Chung, W.K.; Kaltman, J.R.; Miller, T.A. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J. Am. Heart Assoc. 2018, 7, e006906. [Google Scholar] [CrossRef] [PubMed]
- Garg, V. Notch Signaling in Aortic Valve Development and Disease. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; Chapter 53. [Google Scholar]
- Wu, B.; Wang, Y.; Xiao, F.; Butcher, J.T.; Yutzey, K.E.; Zhou, B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annu. Rev. Physiol. 2017, 79, 21–41. [Google Scholar] [CrossRef]
- Muntean, I.; Togănel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, X.J.; Wang, H.J.; Li, W.C.; Chen, L.; Ma, D.; Huang, G.Y. Expression of Cx43-related microRNAs in patients with tetralogy of Fallot. World J. Pediatr. 2014, 10, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef]
- Franco, D.; Campione, M. The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc. Med. 2003, 13, 157–163. [Google Scholar] [CrossRef]
- Hoogaars, W.M.; Barnett, P.; Moorman, A.F.; Christoffels, V.M. T-box factors determine cardiac design. Cell. Mol. Life Sci. 2007, 64, 646–660. [Google Scholar] [CrossRef]
- Kitamura, K.; Miura, H.; Miyagawa-Tomita, S.; Yanazawa, M.; Katoh-Fukui, Y.; Suzuki, R.; Ohuchi, H.; Suehiro, A.; Motegi, Y.; Nakahara, Y.; et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 1999, 126, 5749–5758. [Google Scholar] [PubMed]
- Baldini, A. DiGeorge syndrome: The use of model organisms to dissect complex genetics. Hum. Mol. Genet. 2002, 11, 2363–2369. [Google Scholar] [CrossRef]
- Baldini, A. The 22q11.2 deletion syndrome: A gene dosage perspective. Sci. World J. 2006, 6, 1881–1887. [Google Scholar] [CrossRef]
- Lin, S.; Herdt-Losavio, M.; Gensburg, L.; Marshall, E.; Druschel, C. Maternal asthma, asthma medication use, and the risk of congenital heart defects. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 161–168. [Google Scholar] [CrossRef]
- Gorini, F.; Chiappa, E.; Gargani, L.; Picano, E. Potential effects of environmental chemical contamination in congenital heart disease. Pediatr. Cardiol. 2014, 35, 559–568. [Google Scholar] [CrossRef]
- Baldacci, S.; Gorini, F.; Santoro, M.; Pierini, A.; Minichilli, F.; Bianchi, F. Environmental and individual exposure and the risk of congenital anomalies: A review of recent epidemiological evidence. Epidemiol. Prev. 2018, 42 (Suppl. 1), 1–34. [Google Scholar]
- Wong, P.; Denburg, A.; Dave, M.; Levin, L.; Morinis, J.O.; Suleman, S.; Wong, J.; Ford-Jones, E.; Moore, A.M. Early life environment and social determinants of cardiac health in children with congenital heart disease. Paediatr. Child Health 2018, 23, 92–95. [Google Scholar] [CrossRef]
- Cowan, J.R.; Tariq, M.; Shaw, C.; Rao, M.; Belmont, J.W.; Lalani, S.R.; Smolarek, T.A.; Ware, S.M. Copy number variation as a genetic basis for heterotaxy and heterotaxy-spectrum congenital heart defects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150406. [Google Scholar] [CrossRef]
- Xie, H.M.; Werner, P.; Stambolian, D.; Bailey-Wilson, J.E.; Hakonarson, H.; White, P.S.; Taylor, D.M.; Goldmuntz, E. Rare copy number variants in patients with congenital conotruncal heart defects. Birth Defects Res. 2017, 109, 271–295. [Google Scholar] [CrossRef]
- Shanshen, E.; Rosenberg, J.; Van Bergen, A.H. Identification of Novel Congenital Heart Disease Candidate Genes Using Chromosome Microarray. Pediatr. Cardiol. 2018, 39, 148–159. [Google Scholar] [CrossRef]
- Islas, J.F.; Moreno-Cuevas, J.E. A MicroRNA Perspective on Cardiovascular Development and Diseases: An Update. Int. J. Mol. Sci. 2018, 19, 2075. [Google Scholar] [CrossRef]
- Garcia-Padilla, C.; Aranega, A.E.; Franco, D. The role of long non-coding RNAs in cardiac development and disease. AIMS Genet. 2018, 5, 124–140. [Google Scholar] [CrossRef]
- Rynkeviciene, R.; Simiene, J.; Strainiene, E.; Stankevicius, V.; Usinskiene, J.; Miseikyte Kaubriene, E.; Meskinyte, I.; Cicenas, J.; Suziedelis, K. Non-Coding RNAs in Glioma. Cancers 2018, 11, 17. [Google Scholar] [CrossRef]
- Yamamoto, T.; Saitoh, N. Non-coding RNAs and chromatin domains. Curr. Opin. Cell Biol. 2019, 58, 26–33. [Google Scholar] [CrossRef]
- Expósito-Villén, A.; Aránega, A.; Franco, D. Functional Role of Non-Coding RNAs during Epithelial-To-Mesenchymal Transition. Noncoding RNA 2018, 4, 14. [Google Scholar] [CrossRef]
- Fico, A.; Fiorenzano, A.; Pascale, E.; Patriarca, E.J.; Minchiotti, G. Long non-coding RNA in stem cell pluripotency and lineage commitment: Functions and evolutionary conservation. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, W.; Zhang, B.; Wu, Y.; Yan, S.; Yuan, Y.; Zhang, H.; Li, J.; Sun, K.; Wang, H.; et al. The MALAT1 gene polymorphism and its relationship with the onset of congenital heart disease in Chinese. Biosci. Rep. 2018, 38, BSR20171381. [Google Scholar] [CrossRef]
- Saeedi Borujeni, M.J.; Esfandiary, E.; Baradaran, A.; Valiani, A.; Ghanadian, M.; Codoñer-Franch, P.; Basirat, R.; Alonso-Iglesias, E.; Mirzaei, H.; Yazdani, A. Molecular aspects of pancreatic β-cell dysfunction: Oxidative stress, microRNA, and long noncoding RNA. J. Cell. Physiol. 2019, 234, 8411–8425. [Google Scholar] [CrossRef]
- Zhao, C.X.; Zhu, W.; Ba, Z.Q.; Xu, H.J.; Liu, W.D.; Zhu, B.; Wang, L.; Song, Y.J.; Yuan, S.; Ren, C.P. The regulatory network of nasopharyngeal carcinoma metastasis with a focus on EBV, lncRNAs and miRNAs. Am. J. Cancer Res. 2018, 8, 2185–2209. [Google Scholar]
- Ambros, V. The functions of Animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Hoelscher, S.C.; Doppler, S.A.; Dreßen, M.; Lahm, H.; Lange, R.; Krane, M. MicroRNAs: Pleiotropic players in congenital heart disease and regeneration. J. Thorac. Dis. 2017, 9 (Suppl. 1), S64–S81. [Google Scholar] [CrossRef]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef]
- van Rooij, E.; Olson, E.N. MicroRNAs: Powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Investig. 2007, 117, 2369–2376. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Tahara, N.; Kawakami, Y.; Das, S.; Koyano-Nakagawa, N.; Gong, W.; Garry, M.G.; Garry, D.J. Etv2-miR-130a-Jarid2 cascade regulates vascular patterning during embryogenesis. PLoS ONE 2017, 12, e0189010. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hwangbo, C.; Jaba, I.M.; Zhang, J.; Papangeli, I.; Han, J.; Mikush, N.; Larrivée, B.; Eichmann, A.; Chun, H.J.; et al. miR-182 Modulates Myocardial Hypertrophic Response Induced by Angiogenesis in Heart. Sci. Rep. 2016, 6, 21228. [Google Scholar] [CrossRef]
- Wei, Y.; Peng, S.; Wu, M.; Sachidanandam, R.; Tu, Z.; Zhang, S.; Falce, C.; Sobie, E.A.; Lebeche, D.; Zhao, Y. Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell Res. 2014, 24, 278–292. [Google Scholar] [CrossRef]
- Danielson, L.S.; Park, D.S.; Rotllan, N.; Chamorro-Jorganes, A.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.K.; Hernando, E. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Han, S.; Hu, P.; Zhu, C.; Wang, X.; Qian, L.; Guo, X. Potential role of maternal serum microRNAs as a biomarker for fetal congenital heart defects. Med. Hypotheses 2011, 76, 424–426. [Google Scholar] [CrossRef]
- Zhu, S.; Cao, L.; Zhu, J.; Kong, L.; Jin, J.; Qian, L.; Zhu, C.; Hu, X.; Li, M.; Guo, X.; et al. Identification of maternal serum microRNAs as novel non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Clin. Chim. Acta 2013, 424, 66–72. [Google Scholar] [CrossRef]
- Lai, C.T.; Ng, E.K.; Chow, P.C.; Kwong, A.; Cheung, Y.F. Circulating microRNA expression profile and systemic right ventricular function in adults after atrial switch operation for complete transposition of the great arteries. BMC Cardiovasc. Disord. 2013, 13, 73. [Google Scholar] [CrossRef]
- Tutarel, O.; Dangwal, S.; Bretthauer, J.; Westhoff-Bleck, M.; Roentgen, P.; Anker, S.D.; Bauersachs, J.; Thum, T. Circulating miR-423_5p fails as a biomarker for systemic ventricular function in adults after atrial repair for transposition of the great arteries. Int. J. Cardiol. 2013, 167, 63–66. [Google Scholar] [CrossRef]
- Perry, L.W.; Neill, C.A.; Ferencz, C. EUROCAT Working Party on Congenital Heart Disease: Perspective in Pediatric Cardiology. In Epidemiology of congenital heart disease, the Baltimore-Washington Infant Study 1981–1989; Futura: Armonk, NY, USA, 1993; pp. 33–62. [Google Scholar]
- Becker, A.E.; Connor, M.; Anderson, R.H. Tetralogy of Fallot: A morphometric and geometric study. Am. J. Cardiol. 1975, 35, 402–412. [Google Scholar] [CrossRef]
- Low, K.J.; Buxton, C.C.; Newbury-Ecob, R.A. Tetralogy of Fallot, microcephaly, short stature and brachymesophalangy is associated with hemizygous loss of noncoding MIR17HG and coding GPC5. Clin. Dysmorphol. 2015, 24, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.B.; Mei, J.; Jiang, L.Y.; Jiang, Z.L.; Liu, H.; Zhang, J.W.; Ding, F.B. MiR-196a2 rs11614913 T>C Polymorphism is Associated with an Increased Risk of Tetralogy of Fallot in a Chinese Population. Acta Cardiol. Sin. 2015, 31, 18–23. [Google Scholar]
- O’Brien, J.E., Jr.; Kibiryeva, N.; Zhou, X.G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar] [CrossRef]
- Zhang, J.; Chang, J.J.; Xu, F.; Ma, X.J.; Wu, Y.; Li, W.C.; Wang, H.J.; Huang, G.Y.; Ma, D. MicroRNA deregulation in right ventricular outflow tract myocardium in nonsyndromic tetralogy of fallot. Can. J. Cardiol. 2013, 29, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Bittel, D.C.; Kibiryeva, N.; Marshall, J.A.; O’Brien, J.E., Jr. MicroRNA-421 Dysregulation is Associated with Tetralogy of Fallot. Cells 2014, 3, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, K.; Li, Y.; Shi, K.; Liu, Y.L.; Yang, Y.F.; Fang, Y.; Mao, M. Screening miRNA and their target genes related to tetralogy of Fallot with microarray. Cardiol. Young 2014, 24, 442–446. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Wu, C.; Pan, Z.; Xiang, L.; Liu, H.; Jin, X.; Tong, K.; Fan, S.; Jin, X. Potential association of long noncoding RNA HA117 with tetralogy of Fallot. Genes Dis. 2018, 5, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.T.M.; Ng, E.K.O.; Chow, P.C.; Kwong, A.; Cheung, Y.F. Circulating MicroRNA in patients with repaired tetralogy of Fallot. Eur. J. Clin. Investig. 2017, 47, 574–582. [Google Scholar] [CrossRef]
- Abu-Halima, M.; Meese, E.; Keller, A.; Abdul-Khaliq, H.; Rädle-Hurst, T. Analysis of circulating microRNAs in patients with repaired Tetralogy of Fallot with and without heart failure. J. Transl. Med. 2017, 15, 156. [Google Scholar] [CrossRef]
- Liang, D.; Xu, X.; Deng, F.; Feng, J.; Zhang, H.; Liu, Y.; Zhang, Y.; Pan, L.; Liu, Y.; Zhang, D.; et al. miRNA-940 reduction contributes to human Tetralogy of Fallot development. J. Cell. Mol. Med. 2014, 18, 1830–1839. [Google Scholar] [CrossRef]
- Wilkinson, J.L. Double Outlet Ventricle. In Paedriatric Cardiology; Anderson, R.H., Baker, E.J., Macartney, F.J., Rigb y, M.L., Shinebourne, E.A., Tynan, M., Eds.; Churchill Livingstone: London, UK, 2002; pp. 1353–1382. [Google Scholar]
- Saxena, A.; Tabin, C.J. miRNA-processing enzyme Dicer is necessary for cardiac outflow tract alignment and chamber septation. Proc. Natl. Acad. Sci. USA 2010, 107, 87–91. [Google Scholar] [CrossRef]
- Huang, Z.P.; Chen, J.F.; Regan, J.N.; Maguire, C.T.; Tang, R.H.; Dong, X.R.; Majesky, M.W.; Wang, D.Z. Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2575–2586. [Google Scholar] [CrossRef]
- Belfort, M.A.; Morris, S.A.; Espinoza, J.; Shamshirsaz, A.A.; Sanz Cortez, M.; Justino, H.; Ayres, N.A.; Qureshi, A.M. Thulium laser assisted atrial septal stent placement: First use in fetal hypoplastic left heart syndrome (HLHS) and intact atrial septum. Ultrasound. Obstet. Gynecol. 2018, 24. [Google Scholar] [CrossRef]
- Yabrodi, M.; Mastropietro, C.W. Hypoplastic left heart syndrome: From comfort care to long-term survival. Pediatr. Res. 2017, 81, 142–149. [Google Scholar] [CrossRef]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA expression in hypoplastic left heart syndrome. J. Card. Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Mureşan, D.; Mărginean, C.; Zaharie, G.; Stamatian, F.; Rotar, I.C. Complete atrioventricular septal defect in the era of prenatal diagnosis. Med. Ultrason. 2016, 18, 500–507. [Google Scholar] [CrossRef]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef]
- Le Gloan, L.; Legendre, A.; Iserin, L.; Ladouceur, M. Pathophysiology and natural history of atrial septal defect. J. Thorac. Dis. 2018, 10 (Suppl. 24), S2854–S2863. [Google Scholar] [CrossRef]
- Wiktor, D.M.; Carroll, J.D. ASD Closure in Structural Heart Disease. Curr. Cardiol. Rep. 2018, 20, 37. [Google Scholar] [CrossRef]
- Therrien, J.; Rambihar, S.; Newman, B.; Siminovitch, K.; Langleben, D.; Webb, G.; Granton, J. Eisenmenger syndrome and atrial septal defect: Nature or nurture? Can. J. Cardiol. 2006, 22, 1133–1136. [Google Scholar] [CrossRef]
- Berger, R.M.; Beghetti, M.; Galiè, N.; Gatzoulis, M.A.; Granton, J.; Lauer, A.; Chiossi, E.; Landzberg, M. Atrial septal defects versus ventricular septal defects in BREATHE-5, a placebo-controlled study of pulmonary arterial hypertension related to Eisenmenger’s syndrome: A subgroup analysis. Int. J. Cardiol. 2010, 144, 373–378. [Google Scholar] [CrossRef]
- Song, Y.; Higgins, H.; Guo, J.; Harrison, K.; Schultz, E.N.; Hales, B.J.; Moses, E.K.; Goldblatt, J.; Pachter, N.; Zhang, G. Clinical significance of circulating microRNAs as markers in detecting and predicting congenital heart defects in children. J. Transl. Med. 2018, 16, 42. [Google Scholar] [CrossRef]
- Chen, W.; Li, S. Circulating microRNA as a Novel Biomarker for Pulmonary Arterial Hypertension Due to Congenital Heart Disease. Pediatr. Cardiol. 2017, 38, 86–94. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Song, X.; Liu, L.; Su, G.; Cui, Y. Bioinformatic Analysis of Genes and MicroRNAs Associated with Atrioventricular Septal Defect in Down Syndrome Patients. Int. Heart J. 2016, 57, 490–495. [Google Scholar] [CrossRef]
- Yu, K.; Ji, Y.; Wang, H.; Xuan, Q.K.; Li, B.B.; Xiao, J.J.; Sun, W.; Kong, X.Q. Association of miR-196a2, miR-27a, and miR-499 polymorphisms with isolated congenital heart disease in a Chinese population. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Wang, Y.; Du, X.; Zhou, Z.; Jiang, J.; Zhang, Z.; Ye, L.; Hong, H. A gain-of-function ACTC1 3’UTR mutation that introduces a miR-139-5p target site may be associated with a dominant familial atrial septal defect. Sci. Rep. 2016, 6, 25404. [Google Scholar] [CrossRef]
- Nora, J.J. Multifactorial inheritance hypothesis for the etiology ofcongenital heart diseases: The genetic-environmental interaction. Circulation 1968, 38, 604–617. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge—A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young, endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Li, J.; Cao, Y.; Ma, X.J.; Wang, H.J.; Zhang, J.; Luo, X.; Chen, W.; Wu, Y.; Meng, Y.; Zhang, J.; et al. Roles of miR-1-1 and miR-181c in ventricular septal defects. Int. J. Cardiol. 2013, 168, 1441–1446. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Smith, T.; Rajakaruna, C.; Caputo, M.; Emanueli, C. MicroRNAs in congenital heart disease. Ann. Transl. Med. 2015, 3, 333. [Google Scholar]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef]
- Meder, B.; Katus, H.A.; Rottbauer, W. Right into the heart of microRNA-133a. Genes Dev. 2008, 22, 3227–3231. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Rindt, H.; Gulick, J.; Knotts, S.; Neumann, J.; Robbins, J. In vivo analysis of the murine beta-myosin heavy chain gene promoter. J. Biol. Chem. 1993, 268, 5332–5338. [Google Scholar]
- Chapnik, E.; Sasson, V.; Blelloch, R.; Hornstein, E. Dgcr8 controls neural crest cells survival in cardiovascular development. Dev. Biol. 2012, 362, 50–56. [Google Scholar] [CrossRef]
- Gu, M.; Zheng, A.; Tu, W.; Zhao, J.; Li, L.; Li, M.; Han, S.; Hu, X.; Zhu, J.; Pan, Y.; et al. Circulating LncRNAs as Novel, Non-Invasive Biomarkers for Prenatal Detection of Fetal Congenital Heart Defects. Cell. Physiol. Biochem. 2016, 38, 1459–1471. [Google Scholar] [CrossRef]
- Miranda-Castro, R.; de-Los-Santos-Álvarez, N.; Lobo-Castañón, M.J. Long noncoding RNAs: From genomic junk to rising stars in the early detection of cancer. Anal. Bioanal. Chem. 2019. [Google Scholar] [CrossRef]
- Butova, R.; Vychytilova-Faltejskova, P.; Souckova, A.; Sevcikova, S.; Hajek, R. Long Non-Coding RNAs in Multiple Myeloma. Noncoding RNA 2019, 5, 13. [Google Scholar] [CrossRef]
- Lu, S.; Su, Z.; Fu, W.; Cui, Z.; Jiang, X.; Tai, S. Altered expression of long non-coding RNA GAS5 in digestive tumors. Biosci. Rep. 2019, 39, BSR20180789. [Google Scholar] [CrossRef]
- Harries, L.W. Long non-coding RNAs and human disease. Biochem. Soc. Trans. 2012, 40, 902–906. [Google Scholar] [CrossRef]
- Liu, H.; Song, G.; Zhou, L.; Hu, X.; Liu, M.; Nie, J.; Lu, S.; Wu, X.; Cao, Y.; Tao, L.; et al. Compared analysis of lncRNAs expression profiling in pdk1 gene knockout mice at two time points. Cell. Physiol. Biochem. 2013, 32, 1497–1508. [Google Scholar] [CrossRef]
- Grote, P.; Herrmann, B.G. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Biol. 2013, 10, 1579–1585. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Aguilo, F.; Zhou, M.M.; Walsh, M.J. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res. 2011, 71, 5365–5369. [Google Scholar] [CrossRef]
- Mousavi, K.; Zare, H.; Dell’Orso, S.; Grontved, L.; Gutierrez-Cruz, G.; Derfoul, A.; Hager, G.L.; Sartorelli, V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 2013, 51, 606–617. [Google Scholar] [CrossRef]
- Welsh, I.C.; Kwak, H.; Chen, F.L.; Werner, M.; Shopland, L.S.; Danko, C.G.; Lis, J.T.; Zhang, M.; Martin, J.F.; Kurpios, N.A. Chromatin architecture of the Pitx2 locus requires CTCF-and Pitx2-dependent asymmetry that mirrors embryonic gut laterality. Cell Rep. 2015, 13, 337–349. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef]
- Han, P.; Li, W.; Lin, C.H.; Yang, J.; Shang, C.; Nuernberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.Y.; Lin, C.J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long Noncoding RNAs with snoRNA Ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Furic, L.; Desgroseillers, L.; Maquat, L.E. Mammalian Staufen1 recruits Upf1 to specific mRNA 3’UTRs so as to elicit mRNA decay. Cell 2005, 120, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase expression. Nat. Med. 2008, 14, 723. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef]
- Wang, H.; Iacoangeli, A.; Lin, D.; Williams, K.; Denman, R.B.; Hellen, C.U.; Tiedge, H. Dendritic BC1 RNA in translational control mechanisms. J. Cell Biol. 2005, 171, 811–821. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs:insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol. 2012, 19, 1068–1075. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, Y.; Hu, Y.; Guan, J.; Xu, L.; Xiao, W.; Zhong, Q.; Ren, C.; Lu, J.; Liang, J.; et al. Long noncoding RNA Crnde attenuates cardiac fibrosis via Smad3-Crnde negative feedback in diabetic cardiomyopathy. FEBS J. 2019. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Zhuang, Q.; Pan, R.; Zou, L.; Cen, Z.; Tang, L. Long noncoding RNA homeobox A11 antisense promotes transforming growth factor β1-induced fibrogenesis in cardiac fibroblasts. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, R.; Plaisance, I.; Abraham, B.J.; Sarre, A.; Ting, C.C.; Alexanian, M.; Maric, D.; Maison, D.; Nemir, M.; Young, R.A.; et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017, 9, eaai9118. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Ballarino, M.; Cipriano, A.; Tita, R.; Santini, T.; Desideri, F.; Morlando, M.; Colantoni, A.; Carrieri, C.; Nicoletti, C.; Musarò, A.; et al. Deficiency in the nuclear long noncoding RNA Charme causes myogenic defects and heart remodeling in mice. EMBO J. 2018, 37, e99697. [Google Scholar] [CrossRef]
- Viereck, J.; Kumarswamy, R.; Foinquinos, A.; Xiao, K.; Avramopoulos, P.; Kunz, M.; Dittrich, M.; Maetzig, T.; Zimmer, K.; Remke, J.; et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016, 8, 326ra22. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Liu, F.; Zhang, Y.H.; Li, R.B.; Zhai, M.; Liu, F.; Zhou, L.Y.; Liu, C.Y.; Yan, K.W.; Dong, Y.H.; et al. The Long Non-Coding RNA CPR Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2019. [Google Scholar] [CrossRef]
- Wang, J.; Chen, X.; Shen, D.; Ge, D.; Chen, J.; Pei, J.; Li, Y.; Yue, Z.; Feng, J.; Chu, M.; et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell. Cardiol. 2019, 127, 105–114. [Google Scholar] [CrossRef]
- Chen, G.; Li, H.; Li, X.; Li, B.; Zhong, L.; Huang, S.; Zheng, H.; Li, M.; Jin, G.; Liao, W.; et al. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J. Mol. Cell. Cardiol. 2018, 122, 152–164. [Google Scholar] [CrossRef]
- Song, G.; Shen, Y.; Ruan, Z.; Li, X.; Chen, Y.; Yuan, W.; Ding, X.; Zhu, L.; Qian, L. LncRNA-uc.167 influences cell proliferation, apoptosis and differentiation of P19 cells by regulating Mef2c. Gene 2016, 590, 97–108. [Google Scholar] [CrossRef]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef]
- Hou, J.; Long, H.; Zhou, C.; Zheng, S.; Wu, H.; Guo, T.; Wu, Q.; Zhong, T.; Wang, T. Long noncoding RNA Braveheart promotes cardiogenic differentiation of mesenchymal stem cells in vitro. Stem Cell Res. Ther. 2017, 8, 4. [Google Scholar] [CrossRef]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef]
- Ounzain, S.; Micheletti, R.; Arnan, C.; Plaisance, I.; Cecchi, D.; Schroen, B.; Reverter, F.; Alexanian, M.; Gonzales, C.; Ng, S.Y.; et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J. Mol. Cell. Cardiol. 2015, 89 Pt A, 98–112. [Google Scholar] [CrossRef]
- Anderson, K.M.; Anderson, D.M.; McAnally, J.R.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2016, 539, 433–436. [Google Scholar] [CrossRef]
- Kurian, L.; Aguirre, A.; Sancho-Martinez, I.; Benner, C.; Hishida, T.; Nguyen, T.B.; Reddy, P.; Nivet, E.; Krause, M.N.; Nelles, D.A.; et al. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation 2015, 131, 1278–1290. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhuang, J.; Lin, Y.; Wang, X.; Chen, J.; Han, F. Long noncoding RNA SNHG6 contributes to ventricular septal defect formation via negative regulation of miR-101 and activation of Wnt/β-catenin pathway. Pharmazie 2019, 74, 23–28. [Google Scholar]
- Song, G.; Shen, Y.; Zhu, J.; Liu, H.; Liu, M.; Shen, Y.Q.; Zhu, S.; Kong, X.; Yu, Z.; Qian, L. Integrated analysis of dysregulated lncRNA expression in fetal cardiac tissues with ventricular septal defect. PLoS ONE 2013, 8, e77492. [Google Scholar] [CrossRef]
- Wang, B.; Shi, G.; Zhu, Z.; Chen, H.; Fu, Q. Sexual difference of small RNA expression in Tetralogy of Fallot. Sci. Rep. 2018, 8, 12847. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dueñas, A.; Expósito, A.; Aranega, A.; Franco, D. The Role of Non-Coding RNA in Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2019, 6, 15. https://doi.org/10.3390/jcdd6020015
Dueñas A, Expósito A, Aranega A, Franco D. The Role of Non-Coding RNA in Congenital Heart Diseases. Journal of Cardiovascular Development and Disease. 2019; 6(2):15. https://doi.org/10.3390/jcdd6020015
Chicago/Turabian StyleDueñas, Angel, Almudena Expósito, Amelia Aranega, and Diego Franco. 2019. "The Role of Non-Coding RNA in Congenital Heart Diseases" Journal of Cardiovascular Development and Disease 6, no. 2: 15. https://doi.org/10.3390/jcdd6020015
APA StyleDueñas, A., Expósito, A., Aranega, A., & Franco, D. (2019). The Role of Non-Coding RNA in Congenital Heart Diseases. Journal of Cardiovascular Development and Disease, 6(2), 15. https://doi.org/10.3390/jcdd6020015