
Genetic Architecture of Ischemic Stroke: Insights from Genome-Wide Association Studies and Beyond


Abstract
1. Introduction
2. Genetic Epidemiology of Ischemic Stroke
2.1. Heritability Estimates and Family-Based Studies
2.2. Genetic vs. Environmental Contributions to Ischemic Stroke Risk
2.3. Interaction Between Genetic Risk and Lifestyle Factors
2.4. Stroke Subtypes and Their Genetic Distinctions
3. Genome-Wide Association Studies in Ischemic Stroke
3.1. Antisense Noncoding RNA in the INK4 Locus
3.2. SORT1
3.3. Histone Deacetylase 9
3.4. PITX2 Gene
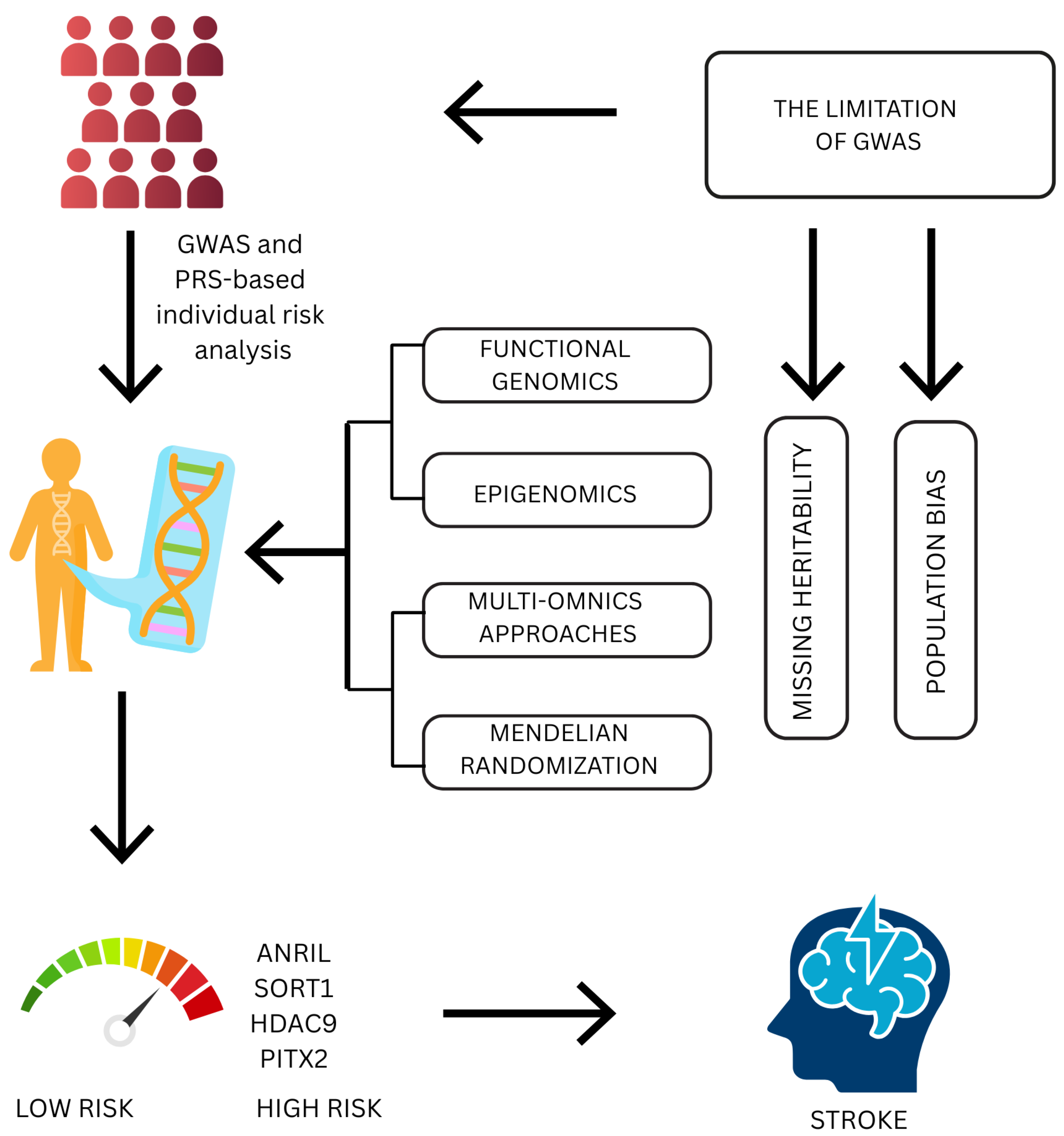
4. Polygenic Risk Scores and Risk Prediction
5. Beyond GWASs: Advanced Genomic Approaches
5.1. Functional Genomics
5.2. Epigenomics
5.3. Multiomics Approaches
5.4. Mendelian Randomization
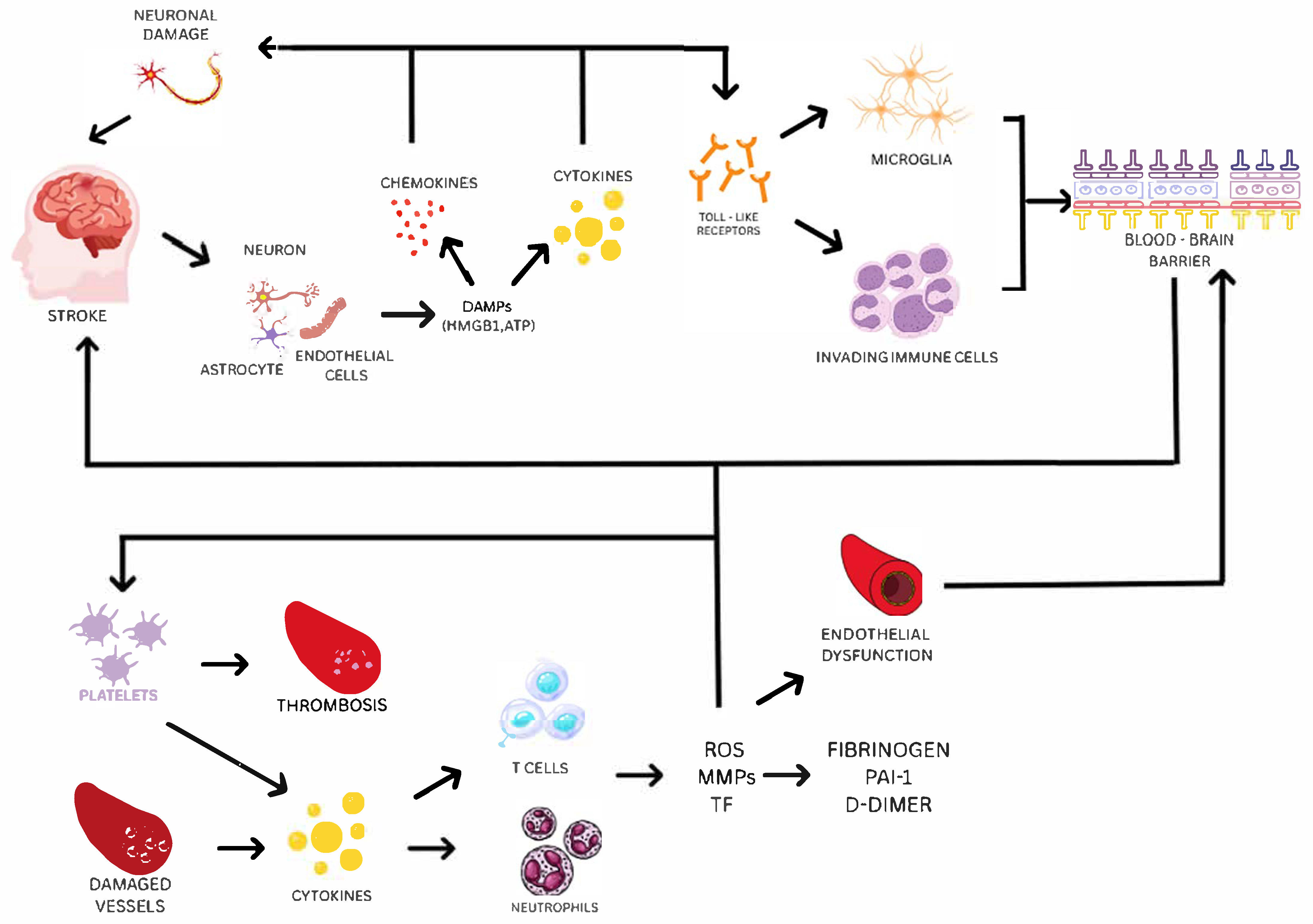
6. Biological Mechanisms and Pathways
6.1. Inflammation, Coagulation, and Endothelial Dysfunction as Key Pathways in Ischemic Stroke
6.2. How GWAS Findings Point to New Insights in Stroke Biology
6.3. Key Genes and Their Involvement in Stroke Risk Pathways
6.3.1. PCSK9
6.3.2. APOE
6.3.3. ARHGEF10
6.3.4. COL4A1/2
6.3.5. IL6-R
6.3.6. F11
7. Clinical Implications and Applications
8. Challenges and Future Directions
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lui, F.; Hui, C.; Khan Suheb, M.Z.; Patti, L. Ischemic Stroke. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2025. [Google Scholar]
- Pu, L.; Wang, L.; Zhang, R.; Zhao, T.; Jiang, Y.; Han, L. Projected global trends in ischemic stroke incidence, deaths and Disability-Adjusted life years from 2020 to 2030. Stroke 2023, 54, 1330–1339. [Google Scholar] [CrossRef]
- Langanay, L.; Gonzalez Sanchez, R.; Hamroun, A.; Dauchet, L.; Amouyel, P.; Dallongeville, J.; Meirhaeghe, A.; Gauthier, V. Ischemic stroke subtypes: Risk factors, treatments, and 1-month prognosis—The Lille, France Stroke Registry. J. Stroke Cerebrovasc. Dis. 2024, 33, 107761. [Google Scholar] [CrossRef]
- Zhang, K.; Loong, S.S.E.; Yuen, L.Z.H.; Venketasubramanian, N.; Chin, H.L.; Lai, P.S.; Tan, B.Y.Q. Genetics in Ischemic Stroke: Current Perspectives and Future Directions. J. Cardiovasc. Dev. Dis. 2023, 10, 495. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Yamagami, H.; Matsumaru, Y. Recent Advances in Stroke Genetics—Unraveling the Complexity of Cerebral Infarction: A Brief Review. Genes 2025, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Sekar, P.K.C.; Veerabathiran, R. A Genetic Perspective on Ischemic Stroke: Recent advances and future directions. Ann. Clin. Exp. Neurol. 2025, 18, 55–67. [Google Scholar] [CrossRef]
- Nikolić, S.; Ignatov, D.I.; Khvorykh, G.V.; Limborska, S.A.; Khrunin, A.V. Genome-wide association studies of ischemic stroke based on interpretable machine learning. PeerJ Comput. Sci. 2024, 10, e2454. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.; de Medeiros Vieira, C.; Mosquera, J.V.; Zade, R.; Parikh, P.; Suryavanshi, S.; Miller, C.L.; Lo Sardo, V. The 9p21.3 Coronary Artery Disease Risk Locus Drives Vascular Smooth Muscle Cells to an Osteochondrogenic State. Arterioscler. Thromb. Vasc. Biol. 2025, 45, 702–721. [Google Scholar] [CrossRef]
- Asare, Y.; Yan, G.; Schlegl, C.; Prestel, M.; van der Vorst, E.P.C.; Teunissen, A.J.P.; Aronova, A.; Tosato, F.; Naser, N.; Caputo, J.; et al. A cis-regulatory element controls expression of histone deacetylase 9 to fine-tune inflammasome-dependent chronic inflammation in atherosclerosis. Immunity 2025, 58, 555–567.e9. [Google Scholar] [CrossRef]
- Ghiasvand, T.; Karimi, J.; Khodadadi, I.; Yazdi, A.; Khazaei, S.; Kichi, Z.A.; Hosseini, S.K. Evaluating SORT1 and SESN1 genes expression in peripheral blood mononuclear cells and oxidative stress status in patients with coronary artery disease. BMC Genomic Data 2024, 25, 93. [Google Scholar] [CrossRef]
- Carré, C.; Carluer, J.B.; Chaux, C.; Estoup-Streiff, C.; Roche, N.; Hosy, E.; Mas, A.; Krouk, G. Next-Gen GWAS: Full 2D epistatic interaction maps retrieve part of missing heritability and improve phenotypic prediction. Genome Biol. 2024, 25, 76. [Google Scholar] [CrossRef]
- Pilalis, E.; Zisis, D.; Andrinopoulou, C.; Karamanidou, T.; Antonara, M.; Stavropoulos, T.G.; Chatziioannou, A. Genome-wide functional annotation of variants: A systematic review of state-of-the-art tools, techniques and resources. Front. Pharmacol. 2025, 16, 1474026. [Google Scholar] [CrossRef]
- Cross, B.; Turner, R.; Pirmohamed, M. Polygenic risk scores: An overview from bench to bedside for personalised medicine. Front. Genet. 2022, 13, 1000667. [Google Scholar] [CrossRef]
- Hughes, J.; Shymka, M.; Ng, T.; Phulka, J.S.; Safabakhsh, S.; Laksman, Z. Polygenic Risk Score Implementation into Clinical Practice for Primary Prevention of Cardiometabolic Disease. Genes 2024, 15, 1581. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Lee, S.B.; Kim, T.K. A brief guide to analyzing expression quantitative trait loci. Mol. Cells 2024, 47, 100139. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.M.B.; Dam, V.S.; Guldbrandsen, H.Ø.; Staehr, C.; Pedersen, T.M.; Kalucka, J.M.; Beck, H.C.; Postnov, D.D.; Lin, L.; Matchkov, V.V. Spatial Transcriptomics and Proteomics Profiling After Ischemic Stroke Reperfusion: Insights into Vascular Alterations. Stroke 2025, 56, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Morris-Blanco, K.C.; Chokkalla, A.K.; Arruri, V.; Jeong, S.; Probelsky, S.M.; Vemuganti, R. Epigenetic mechanisms and potential therapeutic targets in stroke. J. Cereb. Blood Flow. Metab. 2022, 42, 2000–2016. [Google Scholar] [CrossRef]
- Eicher, T.; Kinnebrew, G.; Patt, A.; Spencer, K.; Ying, K.; Ma, Q.; Machiraju, R.; Mathé, A.E.A. Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites 2020, 10, 202. [Google Scholar] [CrossRef]
- Daghlas, I.; Gill, D. Leveraging Mendelian randomization to inform drug discovery and development for ischemic stroke. J. Cereb. Blood Flow. Metab. 2024. advance online publication. [Google Scholar] [CrossRef]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Drexler, C.; Macher, S.; Lindenau, I.; Holter, M.; Moritz, M.; Stojakovic, T.; Pieber, T.R.; Schlenke, P.; Amrein, K. High-dose intravenous versus oral iron in blood donors with iron deficiency: The IronWoMan randomized, controlled clinical trial. Clin. Nutr. 2020, 39, 737–745. [Google Scholar] [CrossRef]
- Sharma, P.; Yadav, S.; Meschia, J.F. Genetics of ischemic stroke. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Bevan, S.; Traylor, M.; Adib-Samii, P.; Malik, R.; Paul, N.L.M.; Jackson, C.; Farrall, M.; Rothwell, P.M.; Sudlow, C.; Dichgans, M.; et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke 2012, 43, 3161–3167. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Liao, Y.C.; Lee, Y.C.; Ihara, M.; Choi, J.C. Update on the Epidemiology, Pathogenesis, and Biomarkers of Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. J. Clin. Neurol. 2023, 19, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Oka, F.; Lee, J.H.; Yuzawa, I.; Li, M.; von Bornstaedt, D.; Eikermann-Haerter, K.; Qin, T.; Chung, D.Y.; Sadeghian, H.; Seidel, J.L.; et al. CADASIL mutations sensitize the brain to ischemia via spreading depolarizations and abnormal extracellular potassium homeostasis. J. Clin. Invest. 2022, 132, e149759. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-J.; Chen, C.-H.; Su, M.-W.; Lin, C.-W.; Cheng, Y.-W.; Tang, S.-C.; Jeng, J.-S. Modifiable vascular risk factors contribute to stroke in 1080 NOTCH3 R544C carriers in Taiwan Biobank. Int. J. Stroke 2023, 19, 105–113. [Google Scholar] [CrossRef]
- Said, M.A.; Verweij, N.; van der Harst, P. Associations of Combined Genetic and Lifestyle Risks with Incident Cardiovascular Disease and Diabetes in the UK Biobank Study. JAMA Cardiol. 2018, 3, 693–702. [Google Scholar] [CrossRef]
- Feigin, V.L.; Abate, M.D.; Abate, Y.H.; ElHafeez, S.A.; Abd-Allah, F.; Abdelalim, A.; Abdelkader, A.; Abdelmasseh, M.; Abd-Elsalam, S.; Abdi, P.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024, 23, 973–1003. [Google Scholar] [CrossRef]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.-K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef]
- Tung, H.; Lin, H.; Chen, P.; Lu, T.; Jhan, P.; Chen, J.; Chen, Y.; Wu, C.; Lin, Y.; Hsiao, T. Characterization of familial hypercholesterolemia in Taiwanese ischemic stroke patients. Aging 2021, 13, 19339–19351. [Google Scholar] [CrossRef]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpón, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef]
- Traylor, M.; Persyn, E.; Tomppo, L.; Klasson, S.; Abedi, V.; Bakker, M.K.; Torres, N.; Li, L.; Bell, S.; Rutten-Jacobs, L.; et al. Genetic basis of lacunar stroke: A pooled analysis of individual patient data and genome-wide association studies. Lancet Neurol. 2021, 20, 351–361. [Google Scholar] [CrossRef] [PubMed]
- French, C.R.; Seshadri, S.; Destefano, A.L.; Fornage, M.; Arnold, C.R.; Gage, P.J.; Skarie, J.M.; Dobyns, W.B.; Millen, K.J.; Liu, T.; et al. Mutation of FOXC1FOXC1 and PITX2 induces cerebral small-vessel disease. J. Clin. Investig. 2014, 124, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Nozaki, H.; Kato, T.; Koyama, A.; Sakai, N.; Ando, S.; Kanazawa, M.; Hishikawa, N.; Nishimoto, Y.; Polavarapu, K.; et al. HTRA1-Related Cerebral Small Vessel Disease: A Review of the Literature. Front. Neurol. 2020, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, A.; Šliachtenko, A.; Grigaitė, J.; Burnytė, B.; Utkus, A.; Jatužis, D. Ischemic Stroke Genetics: What Is New and How to Apply It in Clinical Practice? Genes 2021, 13, 48. [Google Scholar] [CrossRef]
- Debette, S.; Paré, G. Stroke Genetics, Genomics, and Precision Medicine. Stroke 2024, 55, 2163–2168. [Google Scholar] [CrossRef]
- Gorlova, O.Y.; Xiao, X.; Tsavachidis, S.; Amos, C.I.; Gorlov, I.P. SNP characteristics and validation success in genome wide association studies. Hum. Genet. 2022, 141, 229–238. [Google Scholar] [CrossRef]
- Chauhan, W.; Fatma, R.; Wahab, A.; Afzal, M. Cataloging the potential SNPs (single nucleotide polymorphisms) associated with quantitative traits, viz. BMI (body mass index), IQ (intelligence quotient) and BP (blood pressure): An updated review. Egypt. J. Med. Hum. Genet. 2022, 23, 57. [Google Scholar] [CrossRef]
- Dey, K.K.; Gazal, S.; van de Geijn, B.; Kim, S.S.; Nasser, J.; Engreitz, J.M.; Price, A.L. SNP-to-gene linking strategies reveal contributions of enhancer-related and candidate master-regulator genes to autoimmune disease. Cell Genom. 2022, 2, 100145. [Google Scholar] [CrossRef]
- Sasano, T.; Ihara, K.; Tanaka, T.; Furukawa, T. Risk stratification of atrial fibrillation and stroke using single nucleotide polymorphism and circulating biomarkers. PLoS ONE 2023, 18, e0292118. [Google Scholar] [CrossRef]
- Shen, C.; Fan, D.; Fu, H.; Zheng, C.; Chen, Y.; Hu, Z. Single nucleotide polymorphisms in the ANGPTL4 gene and the SNP-SNP interactions on the risk of atherosclerotic ischaemic stroke. BMC Neurol. 2021, 21, 108. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Cui, X.; Lyu, J.; Xie, Y. Exploring genetic associations of 3 types of risk factors with ischemic stroke: An integrated bioinformatics study. Stroke 2024, 55, 1619–1628. [Google Scholar] [CrossRef]
- Mishra, A.; Malik, R.; Hachiya, T.; Jürgenson, T.; Namba, S.; Posner, D.C.; Kamanu, F.K.; Koido, M.; Le Grand, Q.; Shi, M.; et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 2022, 611, 115–123. [Google Scholar] [CrossRef]
- Söderholm, M.; Pedersen, A.; Lorentzen, E.; Stanne, T.M.; Bevan, S.; Olsson, M.; Cole, J.W.; Fernandez-Cadenas, I.; Hankey, G.J.; Jimenez-Conde, J.; et al. Genome-wide association meta-analysis of functional outcome after ischemic stroke. Neurology 2019, 92, e1271–e1283. [Google Scholar] [CrossRef]
- Amini, H.; Knepp, B.; Rodriguez, F.; Jickling, G.C.; Hull, H.; Carmona-Mora, P.; Bushnell, C.; Ander, B.P.; Sharp, F.R.; Stamova, B. Early peripheral blood gene expression associated with good and poor 90-day ischemic stroke outcomes. J. Neuroinflamm. 2023, 20, 13. [Google Scholar] [CrossRef]
- Ibanez, L.; Heitsch, L.; Carrera, C.; Farias, F.H.G.; Del Aguila, J.L.; Dhar, R.; Budde, J.; Bergmann, K.; Bradley, J.; Harari, O.; et al. Multi-ancestry GWAS reveals excitotoxicity associated with outcome after ischaemic stroke. Brain 2022, 145, 2394–2406. [Google Scholar] [CrossRef]
- Majumder, S.N.; Hrastelj, J.; Robertson, N.P. Advances in the genetics of stroke risk and recovery. J. Neurol. 2022, 270, 590–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, S.; Zhang, F.; Akhmedov, S.; Weng, J.; Xu, S. Prioritization of lipid metabolism targets for the diagnosis and treatment of cardiovascular diseases. Research 2025, 8, 0618. [Google Scholar] [CrossRef]
- Granata, A.; Kasioulis, I.; Serrano, F.; Cooper, J.D.; Traylor, M.; Sinha, S.; Markus, H.S. The Histone Deacetylase 9 stroke-risk variant promotes apoptosis and inflammation in a human iPSC-derived smooth muscle cells model. Front. Cardiovasc. Med. 2022, 9, 849664. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Su, X.; Lai, M.; Liu, X.; Cheng, Y. Angiopoietin-like protein family-mediated functions in modulating triglyceride metabolism and related metabolic diseases. Front. Biosci. Landmark 2025, 30, 25862. [Google Scholar] [CrossRef] [PubMed]
- Pereira Ciochetti, N.; Lugli-Moraes, B.; Santos da Silva, B.; Rovaris, D.L. Genome-wide association studies: Utility and limitations for research in physiology. J. Physiol. 2023, 601, 2771–2799. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K.; et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef]
- Razeghian-Jahromi, I.; Karimi Akhormeh, A.; Zibaeenezhad, M.J. The role of ANRIL in atherosclerosis. Dis. Markers 2022, 2022, 8859677. [Google Scholar] [CrossRef]
- Badr, E.A.; Elhelbawy, N.G.; Nagy, A.O.; Sultan, A.A.; Elnaidany, S.S. Association between cyclin-dependent kinase inhibitor 2B antisense RNA 1 and zinc finger homeobox 3 gene polymorphisms and COVID-19 severity. BMC Infect. Dis. 2023, 23, 568. [Google Scholar] [CrossRef] [PubMed]
- Razeghian-Jahromi, I.; Zibaeenezhad, M.J.; Karimi Akhormeh, A.; Dara, M. Expression ratio of circular to linear ANRIL in hypertensive patients with coronary artery disease. Sci. Rep. 2022, 12, 1802. [Google Scholar] [CrossRef] [PubMed]
- Ikonnikova, A.; Anisimova, A.; Galkin, S.; Gunchenko, A.; Abdukhalikova, Z.; Filippova, M.; Surzhikov, S.; Selyaeva, L.; Shershov, V.; Zasedatelev, A.; et al. Genetic association study and machine learning to investigate differences in platelet reactivity in patients with acute ischemic stroke treated with aspirin. Biomedicines 2022, 10, 2564. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Shen, H.; Cha, W.; Xia, H.; Liu, L. Predictive value of using plasma long non-coding RNAs ANRIL and HOXA11-AS for in-stent restenosis. Exp. Ther. Med. 2021, 23, 115. [Google Scholar] [CrossRef]
- Huang, T.; Zhao, H.Y.; Zhang, X.B.; Gao, X.L.; Peng, W.P.; Zhou, Y.; Zhao, W.H.; Yang, H.F. LncRNA ANRIL regulates cell proliferation and migration via sponging miR-339-5p and regulating FRS2 expression in atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1956–1969. [Google Scholar]
- Rodríguez-Esparragón, F.; Torres-Mata, L.B.; Cazorla-Rivero, S.E.; Serna Gómez, J.A.; González Martín, J.M.; Cánovas-Molina, Á.; Medina-Suárez, J.A.; González-Hernández, A.N.; Estupiñán-Quintana, L.; Bartolomé-Durán, M.C.; et al. Analysis of ANRIL isoforms and key genes in patients with severe coronary artery disease. Int. J. Mol. Sci. 2023, 24, 16127. [Google Scholar] [CrossRef]
- Zayani, Z.; Hooshmandi, E.; Borhani-Haghighi, A.; Rahimi, M.; Ostovan, V.R.; Fadakar, N.; Tabrizi, R.; Bayat, M.; Hojati, S.S.; Gharbi, N.; et al. Diagnostic potential of lncRNAs-ANRIL and MIAT in the blood of patients with cerebral venous thrombosis. Curr. J. Neurol. 2024, 23, 117–123. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Hong, S.C.; Yang, Q.; Yin, R.X.; Cao, X.L.; Chen, W.X. Association of variants in CELSR2-PSRC1-SORT1 with risk of serum lipid traits, coronary artery disease and ischemic stroke. Int. J. Clin. Exp. Pathol. 2015, 8, 9543–9551. [Google Scholar]
- Şimsek, Z.; Alizade, E.; Abdurahmanova, İ.; Güner, A.; Zehir, R.; Pala, S. Serum sortilin as a predictor of stroke in patients with intermediate carotid artery stenosis. Vascular 2023, 31, 317–324. [Google Scholar] [CrossRef]
- Chu, X.; Liu, R.; Li, C.; Gao, T.; Dong, Y.; Jiang, Y.; Ke, D. The association of plasma sortilin with essential hypertension and subclinical carotid atherosclerosis: A cross-sectional study. Front. Cardiovasc. Med. 2022, 9, 966890. [Google Scholar] [CrossRef]
- Mitok, K.A.; Schueler, K.L.; King, S.M.; Orr, J.; Ryan, K.A.; Keller, M.P.; Krauss, R.M.; Mitchell, B.D.; Shuldiner, A.R.; Attie, A.D. Missense variants in SORT1 are associated with LDL-C in an Amish population. J. Lipid Res. 2023, 64, 100468. [Google Scholar] [CrossRef]
- Guo, Q.; Kawahata, I.; Cheng, A.; Jia, W.; Wang, H.; Fukunaga, K. Fatty Acid-Binding Proteins: Their Roles in Ischemic Stroke and Potential as Drug Targets. Int. J. Mol. Sci. 2022, 23, 9648. [Google Scholar] [CrossRef]
- Asaro, A.; Sinha, R.; Bakun, M.; Kalnytska, O.; Carlo-Spiewok, A.S.; Rubel, T.; Rozeboom, A.; Dadlez, M.; Kaminska, B.; Aronica, E.; et al. ApoE4 disrupts interaction of sortilin with fatty acid-binding protein 7 essential to promote lipid signaling. J. Cell Sci. 2021, 134, jcs258894. [Google Scholar] [CrossRef]
- Mazella, E.; Mendyk, A.M.; Accart, B.; Borsotto, M.; Heurteaux, C.; Bordet, R.; Mazella, J.; Dondaine, T. Serum sortilin-derived propeptide concentrations as markers of depression in chronic stroke. J. Neurol. Sci. 2025, 472, 123459. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Natarajan, R. HDAC9: An Inflammatory Link in Atherosclerosis. Circ. Res. 2020, 127, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Sanguigno, L.; Guida, N.; Anzilotti, S.; Cuomo, O.; Mascolo, L.; Serani, A.; Brancaccio, P.; Pennacchio, G.; Licastro, E.; Pignataro, G.; et al. Stroke by inducing HDAC9-dependent deacetylation of HIF-1 and Sp1, promotes TfR1 transcription and GPX4 reduction, thus determining ferroptotic neuronal death. Int. J. Biol. Sci. 2023, 19, 2695–2710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mo, X.; Wang, A.; Peng, H.; Guo, D.; Zhong, C.; Zhu, Z.; Xu, T.; Zhang, Y. Association of DNA Methylation in Blood Pressure-Related Genes with Ischemic Stroke Risk and Prognosis. Front. Cardiovasc. Med. 2022, 9, 796245. [Google Scholar] [CrossRef]
- Chou, E.L.; Lino Cardenas, C.L.; Chaffin, M.; Arduini, A.D.; Juric, D.; Stone, J.R.; LaMuraglia, G.M.; Eagleton, M.J.; Conrad, M.F.; Isselbacher, E.M.; et al. Vascular smooth muscle cell phenotype switching in carotid atherosclerosis. JVS Vasc. Sci. 2021, 3, 41–47. [Google Scholar] [CrossRef]
- Ärnlöv, J.; Dluzen, D.F.; Nowak, C. Atherosclerotic Aortic Calcification-Associated Polymorphism in HDAC9 and Associations with Mortality, Cardiovascular Disease, and Kidney Disease. iScience 2020, 23, 101253. [Google Scholar] [CrossRef]
- Hu, Y.; Haessler, J.W.; Manansala, R.; Wiggins, K.L.; Moscati, A.; Beiser, A.; Heard-Costa, N.L.; Sarnowski, C.; Raffield, L.M.; Chung, J.; et al. Trans-Omics for Precision Medicine (TOPMed) Stroke Working Group; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Whole-Genome Sequencing Association Analyses of Stroke and Its Subtypes in Ancestrally Diverse Populations from Trans-Omics for Precision Medicine Project. Stroke 2022, 53, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Zhang, D.; Lv, L.; Shi, W.; Song, Z.; Yi, B.; Lai, B.; Chen, Q.; Yang, S.; Hua, P. Bioinformatic gene analysis for potential biomarkers and therapeutic targets of atrial fibrillation-related stroke. J. Transl. Med. 2019, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, X.; Yang, X.; Yang, M.; Wu, Q. Variants rs2200733 and rs6843082 Show Different Associations in Asian and Non-Asian Populations with Ischemic Stroke. Front. Genet. 2022, 13, 905560. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.T.; Wang, M.Y.; Li, C.N.; Yu, H.; Wang, X.W.; Wu, J.H.; Wang, S.Y.; Wang, J.T.; Chen, D.F.; Wu, T.; et al. Beijing da xue xue bao. Yi xue ban = Journal of Peking University. Health sciences. J. Peking Univ. Health Sci. 2022, 54, 412–420. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Anderson, C.D. Polygenic risk scores in the clinic. Neurology 2023, 100, 693–695. [Google Scholar] [CrossRef]
- Abraham, G.; Rutten-Jacobs, L.; Inouye, M. Risk Prediction Using Polygenic Risk Scores for Prevention of Stroke and Other Cardiovascular Diseases. Stroke 2021, 52, 2983–2991. [Google Scholar] [CrossRef]
- Bebo, A.; Jarmul, J.A.; Pletcher, M.J.; Hasbani, N.R.; Couper, D.; Nambi, V.; Ballantyne, C.M.; Fornage, M.; Morrison, A.C.; Avery, C.L.; et al. Coronary heart disease and ischemic stroke polygenic risk scores and atherosclerotic cardiovascular disease in a diverse, population-based cohort study. PLoS ONE 2023, 18, e0285259. [Google Scholar] [CrossRef]
- Neumann, J.T.; Riaz, M.; Bakshi, A.; Polekhina, G.; Thao, L.T.P.; Nelson, M.R.; Woods, R.L.; Abraham, G.; Inouye, M.; Reid, C.M.; et al. Predictive Performance of a Polygenic Risk Score for Incident Ischemic Stroke in a Healthy Older Population. Stroke 2021, 52, 2882–2891. [Google Scholar] [CrossRef]
- Jung, H.; Jung, H.U.; Baek, E.J.; Kwon, S.Y.; Kang, J.O.; Lim, J.E.; Oh, B. Integration of risk factor polygenic risk score with disease polygenic risk score for disease prediction. Commun. Biol. 2024, 7, 180. [Google Scholar] [CrossRef]
- Hur, H.J.; Yang, H.J.; Kim, M.J.; Lee, K.; Jang, D.J.; Kim, M.S.; Park, S. Interaction of energy and sulfur microbial diet and smoking status with polygenic variants associated with lipoprotein metabolism. Front. Nutr. 2023, 10, 1244185. [Google Scholar] [CrossRef]
- Park, S. Interplay between polygenic variants related immune response and lifestyle factors mitigate the chances of stroke in a genome-wide association study. Br. J. Nutr. 2024, 131, 1813–1826. [Google Scholar] [CrossRef]
- Marston, N.A.; Pirruccello, J.P.; Melloni, G.E.M.; Koyama, S.; Kamanu, F.K.; Weng, L.C.; Roselli, C.; Kamatani, Y.; Komuro, I.; Aragam, K.G.; et al. Predictive Utility of a Coronary Artery Disease Polygenic Risk Score in Primary Prevention. JAMA Cardiol. 2023, 8, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abedi, V.; Zand, R. Dissecting Polygenic Etiology of Ischemic Stroke in the Era of Precision Medicine. J. Clin. Med. 2022, 11, 5980. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chaudhary, D.; Griessenauer, C.J.; Carey, D.J.; Zand, R.; Abedi, V. Predicting mortality among ischemic stroke patients using pathways-derived polygenic risk scores. Sci. Rep. 2022, 12, 12358. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Beeston, E.; Greengrass, C.; Riveros-McKay, F.; Debiec, R.; Lawday, D.; Wang, Q.; Budgeon, C.A.; Braund, P.S.; Bramley, R.; et al. Polygenic risk score adds to a clinical risk score in the prediction of cardiovascular disease in a clinical setting. Eur. Heart J. 2024, 45, 3152–3160. [Google Scholar] [CrossRef]
- Huang, S.; Joshi, A.; Shi, Z.; Wei, J.; Tran, H.; Zheng, S.L.; Duggan, D.; Ashworth, A.; Billings, L.; Helfand, B.T.; et al. Combined polygenic scores for ischemic stroke risk factors aid risk assessment of ischemic stroke. Int. J. Cardiol. 2024, 404, 131990. [Google Scholar] [CrossRef]
- Cai, X.; Li, H.; Cao, X.; Ma, X.; Zhu, W.; Xu, L.; Yang, S.; Yu, R.; Huang, P. Integrating transcriptomic and polygenic risk scores to enhance predictive accuracy for ischemic stroke subtypes. Hum. Genet. 2025, 144, 43–54. [Google Scholar] [CrossRef]
- Slunecka, J.L.; van der Zee, M.D.; Beck, J.J.; Johnson, B.N.; Finnicum, C.T.; Pool, R.; Hottenga, J.J.; de Geus, E.J.C.; Ehli, E.A. Implementation and implications for polygenic risk scores in healthcare. Hum. Genom. 2021, 15, 46. [Google Scholar] [CrossRef]
- Alkis, T.; Luo, X.; Wall, K.; Brody, J.; Bartz, T.; Chang, P.P.; Norby, F.L.; Hoogeveen, R.C.; Morrison, A.C.; Ballantyne, C.M.; et al. A polygenic risk score of atrial fibrillation improves prediction of lifetime risk for heart failure. ESC Heart Fail. 2024, 11, 1086–1096. [Google Scholar] [CrossRef]
- Wang, Y.; Tsuo, K.; Kanai, M.; Neale, B.M.; Martin, A.R. Challenges and Opportunities for Developing More Generalizable Polygenic Risk Scores. Annu. Rev. Biomed. Data Sci. 2022, 5, 293–320. [Google Scholar] [CrossRef]
- Temporelli, P.L. Polygenic risk score and age: An extra help in the cardiovascular prevention of the young? Eur. Heart J. Suppl. 2022, 24 (Suppl. I), I181–I185. [Google Scholar] [CrossRef]
- Omidiran, O.; Patel, A.; Usman, S.; Mhatre, I.; Abdelhalim, H.; DeGroat, W.; Narayanan, R.; Singh, K.; Mendhe, D.; Ahmed, Z. GWAS advancements to investigate disease associations and biological mechanisms. Clin. Transl. Discov. 2024, 4, e296. [Google Scholar] [CrossRef] [PubMed]
- Lovegrove, C.E.; Howles, S.A.; Furniss, D.; Holmes, M.V. Causal inference in health and disease: A review of the principles and applications of Mendelian randomization. J. Bone Miner. Res. 2024, 39, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Granata, A. Functional genomics in stroke: Current and future applications of iPSCs and gene editing to dissect the function of risk variants. BMC Cardiovasc. Disord. 2023, 23, 223. [Google Scholar] [CrossRef] [PubMed]
- Nandy, K.; Babu, D.; Rani, S.; Joshi, G.; Ijee, S.; George, A.; Palani, D.; Premkumar, C.; Rajesh, P.; Vijayanand, S.; et al. Efficient gene editing in induced pluripotent stem cells enabled by an inducible adenine base editor with tunable expression. Sci. Rep. 2023, 13, 21953. [Google Scholar] [CrossRef]
- Tokolyi, A.; Persyn, E.; Nath, A.P.; Burnham, K.L.; Marten, J.; Vanderstichele, T.; Tardaguila, M.; Stacey, D.; Farr, B.; Iyer, V.; et al. The contribution of genetic determinants of blood gene expression and splicing to molecular phenotypes and health outcomes. Nat. Genet. 2025, 57, 616–625. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, H. eQTL studies: From bulk tissues to single cells. J. Genet. Genomics 2023, 50, 925–933. [Google Scholar] [CrossRef]
- Castaldi, P.J.; Abood, A.; Farber, C.R.; Sheynkman, G.M. Bridging the splicing gap in human genetics with long-read RNA sequencing: Finding the protein isoform drivers of disease. Hum. Mol. Genet. 2022, 31, R123–R136. [Google Scholar] [CrossRef]
- Zheng, K.; Lin, L.; Jiang, W.; Chen, L.; Zhang, X.; Zhang, Q.; Ren, Y.; Hao, J. Single-cell RNA-seq reveals the transcriptional landscape in ischemic stroke. J. Cereb. Blood Flow. Metab. 2022, 42, 56–73. [Google Scholar] [CrossRef]
- Costa, S.; La Rocca, G.; Cavalieri, V. Epigenetic Regulation of Chromatin Functions by MicroRNAs and Long Noncoding RNAs and Implications in Human Diseases. Biomedicines 2025, 13, 725. [Google Scholar] [CrossRef] [PubMed]
- Hussen, B.M.; Taheri, M.; Yashooa, R.K.; Abdullah, G.H.; Abdullah, S.R.; Kheder, R.K.; Mustafa, S.A. Revolutionizing medicine: Recent developments and future prospects in stem-cell therapy. Int. J. Surg. 2024, 110, 8002–8024. [Google Scholar] [CrossRef] [PubMed]
- Aboud, N.M.A.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism; StatPearls—NCBI Bookshelf: St. Petersburg, FL, USA, 2023; Available online: https://www.ncbi.nlm.nih.gov/books/NBK532999/ (accessed on 1 May 2025).
- Stanzione, R.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Forte, M.; Volpe, M.; Rubattu, S. Pathogenesis of ischemic stroke: Role of epigenetic mechanisms. Genes 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Choi, I.A.; Lee, J. The role of DNA methylation in stroke recovery. Int. J. Mol. Sci. 2022, 23, 10373. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Zhang, S.Y.; Wen, R.; Zhang, T.N.; Yang, N. Role of histone deacetylases and their inhibitors in neurological diseases. Pharmacol. Res. 2024, 208, 107410. [Google Scholar] [CrossRef]
- Bi, F.; Gao, C.; Guo, H. Epigenetic regulation of cardiovascular diseases induced by behavioral and environmental risk factors: Mechanistic, diagnostic, and therapeutic insights. FASEB BioAdvances 2024, 6, 477–502. [Google Scholar] [CrossRef]
- Li, W.; Shao, C.; Zhou, H.; Du, H.; Chen, H.; Wan, H.; He, Y. Multi-omics research strategies in ischemic stroke: A multidimensional perspective. Ageing Res. Rev. 2022, 81, 101730. [Google Scholar] [CrossRef]
- Zhan, C.; Tang, T.; Wu, E.; Zhang, Y.; He, M.; Wu, R.; Bi, C.; Wang, J.; Zhang, Y.; Shen, B. From multi-omics approaches to personalized medicine in myocardial infarction. Front. Cardiovasc. Med. 2023, 10, 1250340. [Google Scholar] [CrossRef]
- Lu, Q.; Yu, A.; Pu, J.; Chen, D.; Zhong, Y.; Bai, D.; Yang, L. Post-stroke cognitive impairment: Exploring molecular mechanisms and omics biomarkers for early identification and intervention. Front. Mol. Neurosci. 2024, 17, 1375973. [Google Scholar] [CrossRef]
- Pan, S.; Yin, L.; Liu, J.; Tong, J.; Wang, Z.; Zhao, J.; Liu, X.; Chen, Y.; Miao, J.; Zhou, Y.; et al. Metabolomics-driven approaches for identifying therapeutic targets in drug discovery. MedComm 2024, 5, e792. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhu, H.; Feng, S.; Zhang, Y.; Xiong, X. Cross-species multi-omics analysis reveals myeloid-driven endothelial oxidative stress in ischemic stroke. Front. Biosci. (Landmark Ed.) 2025, 30, 37429. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Bi, Y.; Yan, C.; Chen, Y. The impact of inflammation and iron metabolism on gene expression alterations in ischemic stroke: A bioinformatics approach. Sci. Rep. 2025, 15, 15233. [Google Scholar] [CrossRef]
- Michalopoulos, I. Special Issue “Advances in Computational Biology and Bioinformatics” | Michalopoulos Lab | Computational Biology and Bioinformatics. Available online: https://www.michalopoulos.net/blog/?p=443 (accessed on 1 May 2025).
- Richmond, R.C.; Davey Smith, G. Mendelian randomization: Concepts and scope. Cold Spring Harb. Perspect. Med. 2022, 12, a040501. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Butterworth, A.S.; Burgess, S. Mendelian randomization for cardiovascular diseases: Principles and applications. Eur. Heart J. 2023, 44, 4913–4924. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Gill, D. Mendelian randomization studies in stroke: Exploration of risk factors and drug targets with human genetic data. Stroke 2021, 52, 2992–3003. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.F.; Finan, C.; Gordillo-Marañón, M.; Asselbergs, F.W.; Freitag, D.F.; Patel, R.S.; Tyl, B.; Chopade, S.; Faraway, R.; Zwierzyna, M.; et al. Genetic drug target validation using Mendelian randomisation. Nat. Commun. 2020, 11, 3255. [Google Scholar] [CrossRef]
- Yang, T.; Xie, W.; Hu, F.; Cai, B. Causal relationship between C-reactive protein and ischemic stroke caused by atherosclerosis: A Mendelian randomization study. J. Stroke Cerebrovasc. Dis. 2024, 33, 107873. [Google Scholar] [CrossRef]
- Sanderson, E.; Glymour, M.M.; Holmes, M.V.; Kang, H.; Morrison, J.; Munafò, M.R.; Palmer, T.; Schooling, C.M.; Wallace, C.; Zhao, Q.; et al. Mendelian randomization. Nat. Rev. Methods Primers 2022, 2, 6. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Luo, Y.; Dong, W.; Yuan, L.; Zhu, Y.A.; Zhang, D.D.; Ni, H.; Zhu, W. The role of thrombo-inflammation in ischemic stroke: Focus on the manipulation and clinical application. Mol. Neurobiol. 2025, 62, 2362–2375. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, S.; Marlborough, F.; Doubal, F.; Webb, D.J.; Wardlaw, J. Blood markers of coagulation, fibrinolysis, endothelial dysfunction and inflammation in lacunar stroke versus non-lacunar stroke and non-stroke: Systematic review and meta-analysis. Cerebrovasc. Dis. 2014, 37, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Daidone, M.; Pinto, A. Endothelial dysfunction and inflammation in ischemic stroke pathogenesis. Curr. Pharm. Des. 2020, 26, 4209–4219. [Google Scholar] [CrossRef]
- Gong, R.; Tan, J.L.; Liu, G.; Liu, X.F.; Ma, L.; Shi, S. Mechanism of disturbed endothelial cell function on angiogenesis following ischemic brain stroke (Review). Exp. Ther. Med. 2025, 29, 61. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Shekhar, S.; Cunningham, M.W.; Pabbidi, M.R.; Wang, S.; Booz, G.W.; Fan, F. Targeting vascular inflammation in ischemic stroke: Recent developments on novel immunomodulatory approaches. Eur. J. Pharmacol. 2018, 833, 531–544. [Google Scholar] [CrossRef]
- Maiocchi, S.; Alwis, I.; Wu, M.C.L.; Yuan, Y.; Jackson, S.P. Thromboinflammatory functions of platelets in ischemia-reperfusion injury and its dysregulation in diabetes. Semin. Thromb. Hemost. 2018, 44, 102–113. [Google Scholar] [CrossRef]
- Traylor, M.; Farrall, M.; Holliday, E.G.; Sudlow, C.; Hopewell, J.C.; Cheng, Y.C.; Fornage, M.; Ikram, M.A.; Malik, R.; Bevan, S.; et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): A meta-analysis of genome-wide association studies. Lancet Neurol. 2012, 11, 951–962. [Google Scholar] [CrossRef]
- Natarajan, P.; Peloso, G.M.; Zekavat, S.M.; Montasser, M.; Ganna, A.; Chaf, M.; Khera, A.V.; Zhou, W.; Bloom, J.M.; Engreitz, J.M.; et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat. Commun. 2018, 9, 3391. [Google Scholar] [CrossRef]
- Yuan, S.; Tang, B.; Zheng, J.; Larsson, S.C. Circulating lipoprotein lipids, apolipoproteins and ischemic stroke. Ann. Neurol. 2020, 88, 1229–1236. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; McQueen, M.; Sniderman, A.; Pare, G.; Wang, X.; Hankey, G.J.; Rangarajan, S.; Chin, S.L.; Rao-Melacini, P.; Ferguson, J.; et al. INTERSTROKE Investigators. Association of lipids, lipoproteins, and apolipoproteins with stroke subtypes in an international case control study (INTERSTROKE). J. Stroke 2022, 24, 224–235. [Google Scholar] [CrossRef]
- Wu, C.; Huang, R.T.; Kuo, C.H.; Kumar, S.; Kim, C.W.; Lin, Y.C.; Chen, Y.J.; Birukova, A.; Birukov, K.G.; Dulin, N.O.; et al. Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ. Res. 2015, 117, e41–e53. [Google Scholar] [CrossRef] [PubMed]
- Chmelova, M.; Androvic, P.; Kirdajova, D.; Tureckova, J.; Kriska, J.; Valihrach, L.; Anderova, M.; Vargova, L. A view of the genetic and proteomic profile of extracellular matrix molecules in aging and stroke. Front. Cell. Neurosci. 2023, 17, 1296455. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.; Traylor, M. Twelve loci provide insights into the genetic basis of lacunar stroke and small vessel disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2021, 20, 351–361. [Google Scholar] [CrossRef]
- Bordes, C.; Sargurupremraj, M.; Mishra, A.; Debette, S. Genetics of common cerebral small vessel disease. Nat. Rev. Neurol. 2022, 18, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.F.; Duconge-Soler, J.; Franqui-Rivera, H.; Feliu-Maldonado, R.; Roche-Lima, A.; Almodovar-Rivera, I. Insight on the genetics of atrial fibrillation in Puerto Rican Hispanics. Stroke Res. Treat. 2021, 2021, 8819896. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Qiao, S.Y.; Shang, K.; Chu, Y.H.; Yu, H.H.; Chen, X.; Qin, C.; Pan, D.J.; Tian, D.S. Apolipoprotein E ε4 polymorphism as a risk factor for ischemic stroke: A systematic review and meta-analysis. Dis. Markers 2022, 2022, 1407183. [Google Scholar] [CrossRef]
- Li, H.; Yu, S.; Wang, R.; Sun, Z.; Zhou, X.; Zheng, L.; Yin, Z.; Zhang, X.; Sun, Y. ARHGEF10 gene polymorphism is closely associated with the risk of ischemic stroke in Northern Han Chinese population. Neurol. Res. 2017, 39, 158–164. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Malik, R.; Gill, D.; Franceschini, N.; Sudlow, C.L.M.; Dichgans, M.; INVENT Consortium; CHARGE Inflammation Working Group. Interleukin-6 signaling effects on ischemic stroke and other cardiovascular outcomes: A Mendelian randomization study. Circ. Genom. Precis. Med. 2020, 13, e002872. [Google Scholar] [CrossRef]
- Daghlas, I.; Karhunen, V.; Kim, A.S.; Gill, D. Application of human genetics to prioritize coagulation cascade protein targets for ischemic stroke prevention. Stroke 2025, 56, 1542–1553. [Google Scholar] [CrossRef]
- Engelen, M.M.; Van Edom, C.; Verstraete, A.; Verhamme, P.; Vanassche, T. The current landscape of factor XI inhibitors. Thromb. Res. 2024, 235, 108118. [Google Scholar] [CrossRef]
- Bragazzi, N.L.; Zhang, L.; Omarov, M.; Georgakis, M.K. Genetic risk scores in stroke research and care. Stroke 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.J.; Maksemous, N.; Smith, R.A.; Sutherland, H.G.; Haupt, L.M.; Griffiths, L.R. Targeted exonic sequencing identifies novel variants in a cerebral small vessel disease cohort. Clin. Chim. Acta 2025, 567, 120120. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Kraemer, M.; Burlina, A.; Mancuso, M.; Finsterer, J.; Sacco, S.; Salvarani, C.; Caputi, L.; Chabriat, H.; Oberstein, S.L.; et al. Heritable and non-heritable uncommon causes of stroke. J. Neurol. 2021, 268, 2780–2807. [Google Scholar] [CrossRef]
- Delabays, B.; Trajanoska, K.; Walonoski, J.; Mooser, V. Cardiovascular pharmacogenetics: From discovery of genetic association to clinical adoption of derived test. Pharmacol. Rev. 2024, 76, 791–827. [Google Scholar] [CrossRef]
- Shubbar, Q.; Alchakee, A.; Issa, K.W.; Adi, A.J.; Shorbagi, A.I.; Saber-Ayad, M. From genes to drugs: CYP2C19 and pharmacogenetics in clinical practice. Front. Pharmacol. 2024, 15, 1326776. [Google Scholar] [CrossRef]
- Sabatello, M.; Bakken, S.; Chung, W.K.; Cohn, E.; Crew, K.D.; Kiryluk, K.; Kukafka, R.; Weng, C.; Appelbaum, P.S. Return of polygenic risk scores in research:Stakeholders’ views on the eMERGE-IV study. HGG Adv. 2024, 5, 100281. [Google Scholar] [CrossRef]
- Jegede, A.; Balogun, O.; Olorunsogbon, O.F.; Nichols, M.; Akinyemi, J.; Jenkins, C.; Ogunronbi, M.; Singh, A.; Obiako, R.; Wahab, K.; et al. Research participants’ perception of ethical issues in stroke genomics and neurobiobanking research in Africa. PLoS ONE 2025, 20, e0292906. [Google Scholar] [CrossRef] [PubMed]
- Fatumo, S.; Chikowore, T.; Choudhury, A.; Ayub, M.; Martin, A.R.; Kuchenbaecker, K. A roadmap to increase diversity in genomic studies. Nat. Med. 2022, 28, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Debette, S.; Markus, H.S. Stroke genetics: Discovery, insight into mechanisms, and clinical perspectives. Circ. Res. 2022, 130, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Llucià-Carol, L.; Muiño, E.; Cullell, N.; Cárcel-Márquez, J.; Lledós, M.; Gallego-Fabrega, C.; Martin-Campos, J.; Martí-Fàbregas, J.; Aguilera-Simón, A.; Planas, A.M.; et al. Genetic architecture of ischaemic strokes after COVID-19 shows similarities with large vessel strokes. Int. J. Mol. Sci. 2023, 24, 13452. [Google Scholar] [CrossRef]
- Xiang, R.; Kelemen, M.; Xu, Y.; Harris, L.W.; Parkinson, H.; Inouye, M.; Lambert, S.A. Recent advances in polygenic scores: Translation, equitability, methods and FAIR tools. Genome Med. 2024, 16, 33. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, M.; Ma, H.; Shen, H. Polygenic risk scores: The future of cancer risk prediction, screening, and precision prevention. Med. Rev. 2022, 1, 129–149. [Google Scholar] [CrossRef]
- Molla, G.; Bitew, M. Revolutionizing personalized medicine: Synergy with multi-omics data generation, main hurdles, and future perspectives. Biomedicines 2024, 12, 2750. [Google Scholar] [CrossRef]
- Williamson, S.M.; Prybutok, V. Balancing privacy and progress: A review of privacy challenges, systemic oversight, and patient perceptions in AI-driven healthcare. Appl. Sci. 2024, 14, 675. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR gene therapy: Applications, limitations, and implications for the future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Olawade, D.B.; Aderinto, N.; Clement David-Olawade, A.; Egbon, E.; Adereni, T.; Popoola, M.R.; Tiwari, R. Integrating AI-driven wearable devices and biometric data into stroke risk assessment: A review of opportunities and challenges. Clin. Neurol. Neurosurg. 2025, 249, 108689. [Google Scholar] [CrossRef]
- Woodman, R.J.; Mangoni, A.A. A comprehensive review of machine learning algorithms and their application in geriatric medicine: Present and future. Aging Clin. Exp. Res. 2023, 35, 2363–2397. [Google Scholar] [CrossRef]
- Nicholls, H.L.; John, C.R.; Watson, D.S.; Munroe, P.B.; Barnes, M.R.; Cabrera, C.P. Reaching the end-game for GWAS: Machine learning approaches for the prioritization of complex disease loci. Front. Genet. 2020, 11, 350. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene | Function/Role | Stroke Association | Mechanism/Molecular Pathway | Reference |
|---|---|---|---|---|
| PHACTR1 | Regulates actin cytoskeleton and endothelial function | Associated with large-artery atherosclerotic stroke | Involved in vascular remodeling and endothelial dysfunction | [29] |
| LDLR | Low-density lipoprotein receptor involved in cholesterol metabolism | Increases risk of large-artery stroke (especially in familial hypercholesterolemia) | Modulates circulating levels of cholesterol; influences atherosclerosis | [30] |
| ZFHX3 | Transcription factor involved in cardiac conduction | Strongly associated with cardioembolic stroke | Implicated in atrial fibrillation and atrial remodeling | [29] |
| PITX2 | Homeobox transcription factor involved in cardiac development | Strong association with cardioembolic stroke | Alters atrial electrophysiology and structure; influences risk of atrial fibrillation | [31] |
| COL4A1/COL4A2 | Encode type IV collagen; key in vascular basement membrane | Associated with lacunar stroke and cerebral microbleeds | Disrupts blood–brain barrier and vessel integrity | [32] |
| FOXC1 | Transcription factor involved in neurovascular development | Linked to white matter lesions and small-vessel disease | Influences brain vasculature and white matter health | [33] |
| HTRA1 | Serine protease; regulator in familial cerebral small-vessel disease | Implicated in both familial and sporadic lacunar stroke | Modulates extracellular matrix and TGF-beta signaling | [34] |
| ANGPTL4 | Angiopoietin-like protein involved in lipid metabolism | Associated with reduced risk of atherosclerotic stroke | Regulates lipid levels and vascular inflammation | [41] |
| FURIN | Protease involved in protein processing and neuroprotection | Implicated in ischemic stroke susceptibility | Involved in lipid metabolism, neuronal repair pathways | [42] |
| ALDH2 | Enzyme involved in oxidative stress response and DNA repair | Associated with ischemic stroke | Detoxifies reactive aldehydes and reduces neuronal injury | [42] |
| TOMM40 | Mitochondrial membrane protein | Associated with ischemic stroke | Involved in neuroprotection and mitochondrial integrity | [42] |
| ATP2B1 | Calcium transport gene | Influences stroke outcome | Regulates vascular tone and blood pressure | [43] |
| GRK5 | G protein-coupled receptor kinase | Linked to ischemic stroke prognosis | Influences cardiovascular remodeling and inflammation | [43] |
| SH3PXD2A | Cell migration and matrix remodeling gene | Implicated in stroke recovery | Modulates extracellular matrix degradation and vascular repair | [43] |
| CENPQ | Centromere protein Q | Associated with stroke recovery outcomes | Regulates cell cycle and genomic stability | [43] |
| HOXC4 | Transcription factor | Linked to stroke prognosis | Regulates developmental genes involved in repair processes | [44] |
| BNC2 | Transcription factor | Associated with stroke susceptibility and outcome | Modulates gene expression related to inflammation | [45] |
| ADAM23 | Involved in neuronal adhesion and excitability | Linked to poor stroke outcome | Influences synaptic function and excitotoxic damage | [46] |
| GRIA1 | Glutamate receptor subunit | Associated with early neurological instability | Mediates excitotoxicity post-stroke | [46] |
| ANRIL | Long noncoding RNA regulating cell proliferation and vascular health | Increases ischemic stroke risk (especially large-artery subtype) | Influences vascular smooth muscle cell growth, inflammation, and atherosclerosis | [53,54,55,56,57,58,59,60] |
| SORT1 | Encodes sortilin; involved in lipoprotein metabolism | Increases ischemic stroke risk | Modulates cholesterol levels, inflammation, and endothelial function | [61,62,63,64,65,66,67] |
| HDAC9 | Histone deacetylase influencing inflammation and vascular remodeling | Strong association with ischemic stroke risk and progression | Activates NF-kappaB; enhances atherosclerosis, ferroptosis, and plaque instability | [68,69,70,71,72] |
| PPAP2B | Encodes lipid phosphate phosphatase 3; endothelial barrier regulator | Associated with ischemic stroke | Maintains blood–brain barrier integrity by degrading lysophosphatidic acid | [136] |
| ARHGEF10 | Rho guanine nucleotide exchange factor | Linked to ischemic stroke risk in Han Chinese population | Alters endothelial permeability via actin cytoskeleton remodeling | [144,145] |
| APOE | Lipid transporter in the brain and vasculature | Increases risk of small-vessel stroke | Impairs lipid clearance and damages blood–brain barrier | [143] |
| PCSK9 | Modulates LDL receptor degradation | Loss of function reduces ischemic stroke risk | Lowers LDL cholesterol; therapeutic target for stroke prevention | [133] |
| IL6-R | Interleukin-6 receptor | Reduced function linked to lower stroke risk | Anti-inflammatory pathway modulated via Asp358Ala variant | [146] |
| F11 | Encodes coagulation factor XI | Increased levels linked to higher cardioembolic stroke risk | Enhances thrombin generation; potential antithrombotic drug target | [147,148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagodic, A.; Zivalj, D.; Krsek, A.; Baticic, L. Genetic Architecture of Ischemic Stroke: Insights from Genome-Wide Association Studies and Beyond. J. Cardiovasc. Dev. Dis. 2025, 12, 281. https://doi.org/10.3390/jcdd12080281
Jagodic A, Zivalj D, Krsek A, Baticic L. Genetic Architecture of Ischemic Stroke: Insights from Genome-Wide Association Studies and Beyond. Journal of Cardiovascular Development and Disease. 2025; 12(8):281. https://doi.org/10.3390/jcdd12080281
Chicago/Turabian StyleJagodic, Ana, Dorotea Zivalj, Antea Krsek, and Lara Baticic. 2025. "Genetic Architecture of Ischemic Stroke: Insights from Genome-Wide Association Studies and Beyond" Journal of Cardiovascular Development and Disease 12, no. 8: 281. https://doi.org/10.3390/jcdd12080281
APA StyleJagodic, A., Zivalj, D., Krsek, A., & Baticic, L. (2025). Genetic Architecture of Ischemic Stroke: Insights from Genome-Wide Association Studies and Beyond. Journal of Cardiovascular Development and Disease, 12(8), 281. https://doi.org/10.3390/jcdd12080281