
Dilated Cardiomyopathy: A Novel BAG3 Mutation Associated with Aggressive Disease Progression and Ventricular Arrhythmias

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
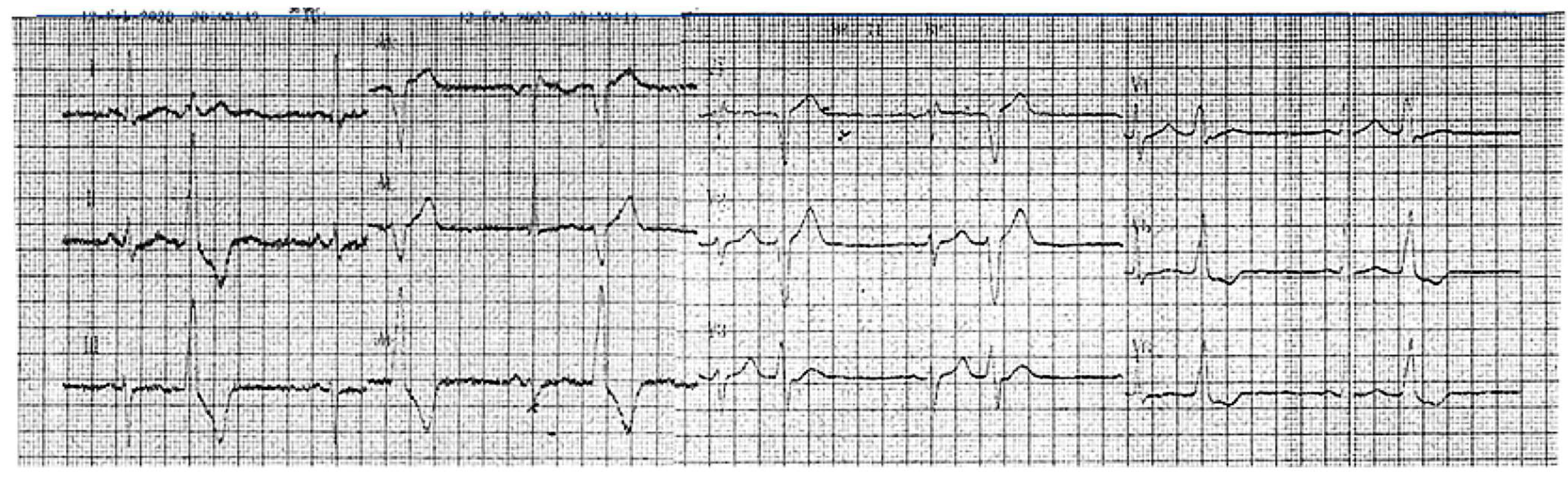
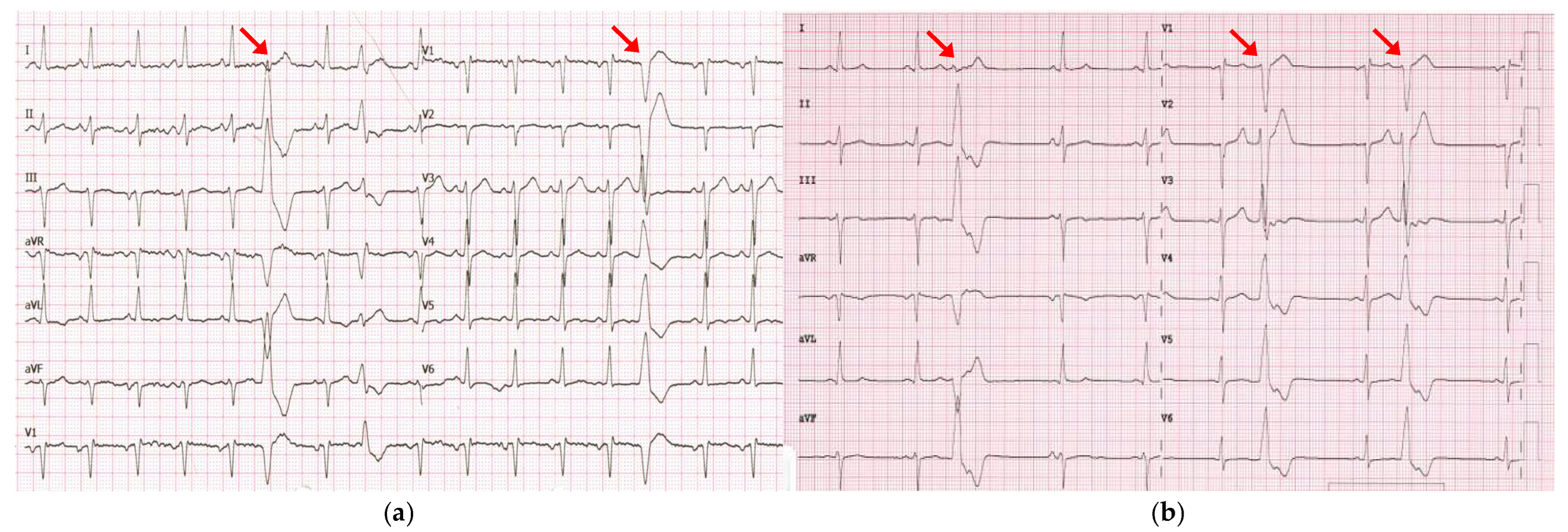
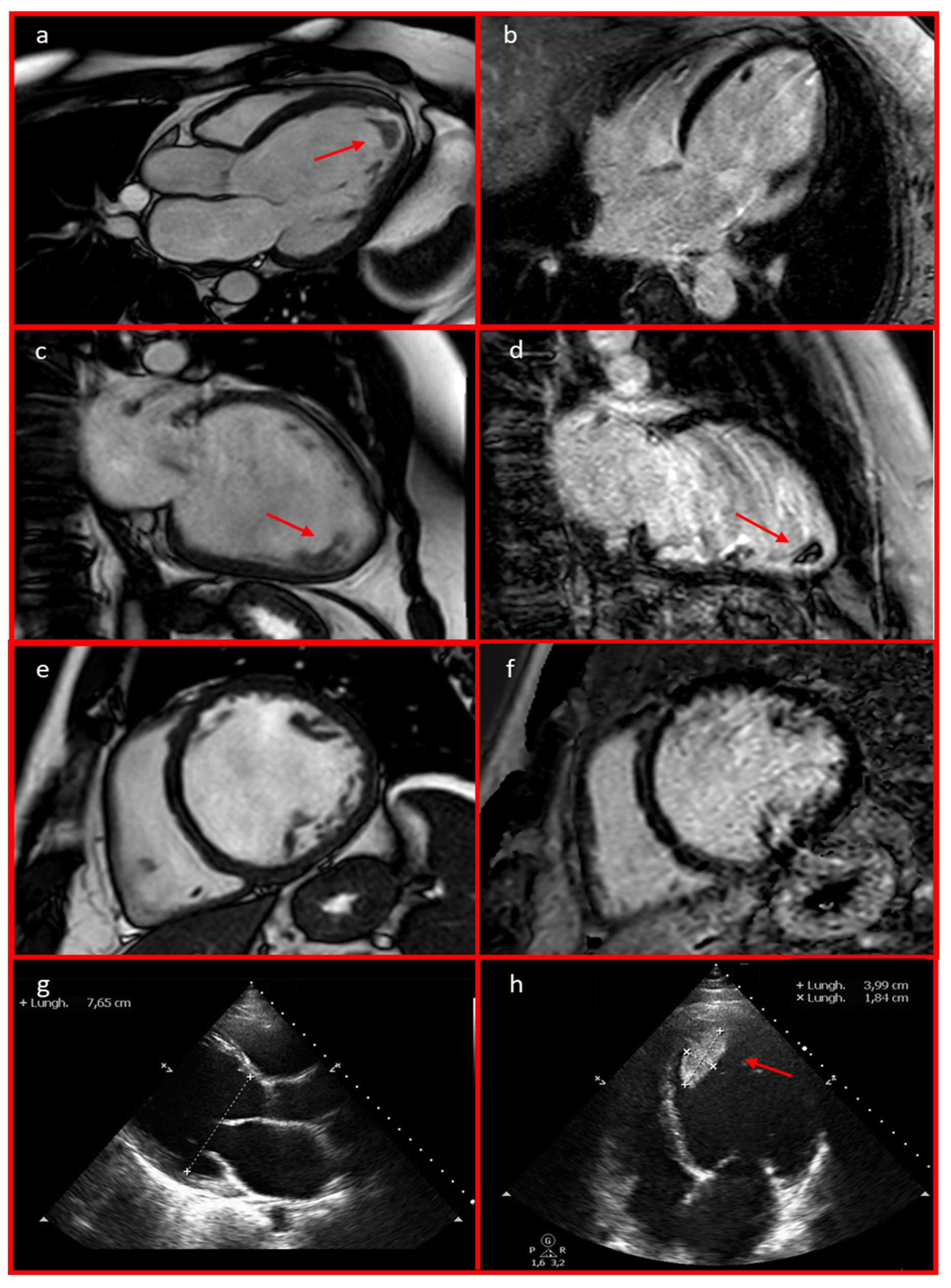
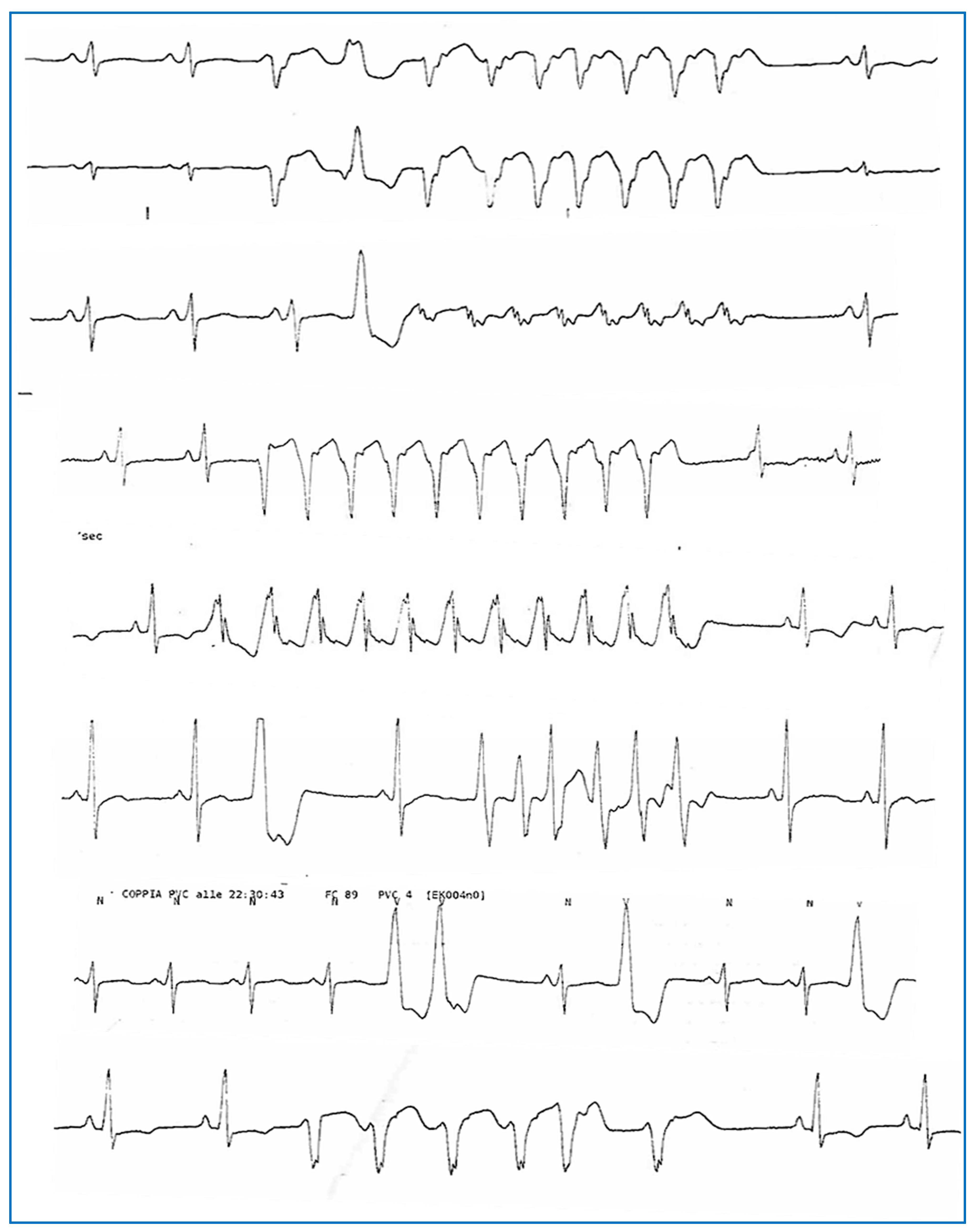
2. Detailed Case Description
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | dilated cardiomyopathy |
| BAG3 | BCL2-associated athanogene 3 |
| HF | heart failure |
| VA | ventricular arrhythmias |
| PVCs | premature ventricular contractions |
| LV | left ventricular |
| EF | ejection fraction |
| ECG | electrocardiogram |
| MRI | magnetic resonance imaging |
| LVEDD | left ventricular end-diastolic diameter |
| NSVT | non-sustained ventricular tachycardia |
| WCD | wearable cardioverter-defibrillator |
| LMNA | Lamin A/C protein-coding gene |
| S-ICD | subcutaneous implantable cardioverter-defibrillator |
References
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated Cardiomyopathy: From Epidemiologic to Genetic Phenotypes: A Translational Review of Current Literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [PubMed]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.W.; et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [PubMed]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Züchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [PubMed]
- Liu, L.; Sun, K.; Zhang, X.; Tang, Y.; Xu, D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 183–194. [Google Scholar] [PubMed]
- Domínguez, F.; Cuenca, S.; Bilinska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, T.; Myers, V.D.; Gordon, J.; Tilley, D.J.; Sharp, T.E., 3rd; Wang, J.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. BAG3: A new player in the heart failure paradigm. Heart Fail. Rev. 2015, 20, 423–434. [Google Scholar] [PubMed]
- Myers, V.D.; McClung, J.M.; Wang, J.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Kontos, C.H.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. The Multifunctional Protein BAG3 A Novel Therapeutic Target in Cardiovascular Disease. J. Am. Coll. Cardiol. Basic Trans. Sci. 2018, 3, 122–131. [Google Scholar]
- Feldman, A.M.; Gordon, J.; Wang, J.; Song, J.; Zhang, X.; Myers, V.D.; Tilley, D.J.; Gao, E.; Hoffman, N.E.; Tomar, D.; et al. BAG3 regulates contractility and Ca2+ homeostasis in adult mouse ventricular myocytes. J. Mol. Cell Cardiol. 2016, 92, 10–20. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastori, P.; Balla, C.; Rasia, M.; Lo Jacono, E.; Guerra, C.; Schininà, R.; Gualandi, F.; Bertini, M.; Tortorella, G. Dilated Cardiomyopathy: A Novel BAG3 Mutation Associated with Aggressive Disease Progression and Ventricular Arrhythmias. J. Cardiovasc. Dev. Dis. 2025, 12, 121. https://doi.org/10.3390/jcdd12040121
Pastori P, Balla C, Rasia M, Lo Jacono E, Guerra C, Schininà R, Gualandi F, Bertini M, Tortorella G. Dilated Cardiomyopathy: A Novel BAG3 Mutation Associated with Aggressive Disease Progression and Ventricular Arrhythmias. Journal of Cardiovascular Development and Disease. 2025; 12(4):121. https://doi.org/10.3390/jcdd12040121
Chicago/Turabian StylePastori, Paolo, Cristina Balla, Marta Rasia, Emilia Lo Jacono, Clelia Guerra, Roberta Schininà, Francesca Gualandi, Matteo Bertini, and Giovanni Tortorella. 2025. "Dilated Cardiomyopathy: A Novel BAG3 Mutation Associated with Aggressive Disease Progression and Ventricular Arrhythmias" Journal of Cardiovascular Development and Disease 12, no. 4: 121. https://doi.org/10.3390/jcdd12040121
APA StylePastori, P., Balla, C., Rasia, M., Lo Jacono, E., Guerra, C., Schininà, R., Gualandi, F., Bertini, M., & Tortorella, G. (2025). Dilated Cardiomyopathy: A Novel BAG3 Mutation Associated with Aggressive Disease Progression and Ventricular Arrhythmias. Journal of Cardiovascular Development and Disease, 12(4), 121. https://doi.org/10.3390/jcdd12040121