
Genomic Analysis of Porcine Reproductive and Respiratory Syndrome Virus 1 Revealed Extensive Recombination and Potential Introduction Events in China

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Processing and Sequencing
2.2. Virus Isolation
2.3. Dataset
2.4. Recombination Analysis
2.5. NSP2 Polymorphic Patterns
2.6. Phylogenetic Analysis
2.7. Quantitative Reverse-Transcription PCR (RT-qPCR)
2.7.1. Design of Primers and Probe
2.7.2. RT-qPCR Assay
2.7.3. Generation of the Standard Curve
2.7.4. Sensitivity Analysis, Specificity Analysis, Reproducibility, and Repeatability Analysis
2.8. Nucleotide Sequence Accession Numbers
3. Results
3.1. Identification of PRRSV-1 Samples in Mainland China
3.2. Genomic Sequence Analysis of Three PRRSV-1 Samples
3.3. Deletion and Mutations of the Three PRRSV-1 Samples
3.4. Recombination Analysis of PRRSV-1 Samples
3.5. Comparison of NSP2 at the Amino Acid Level
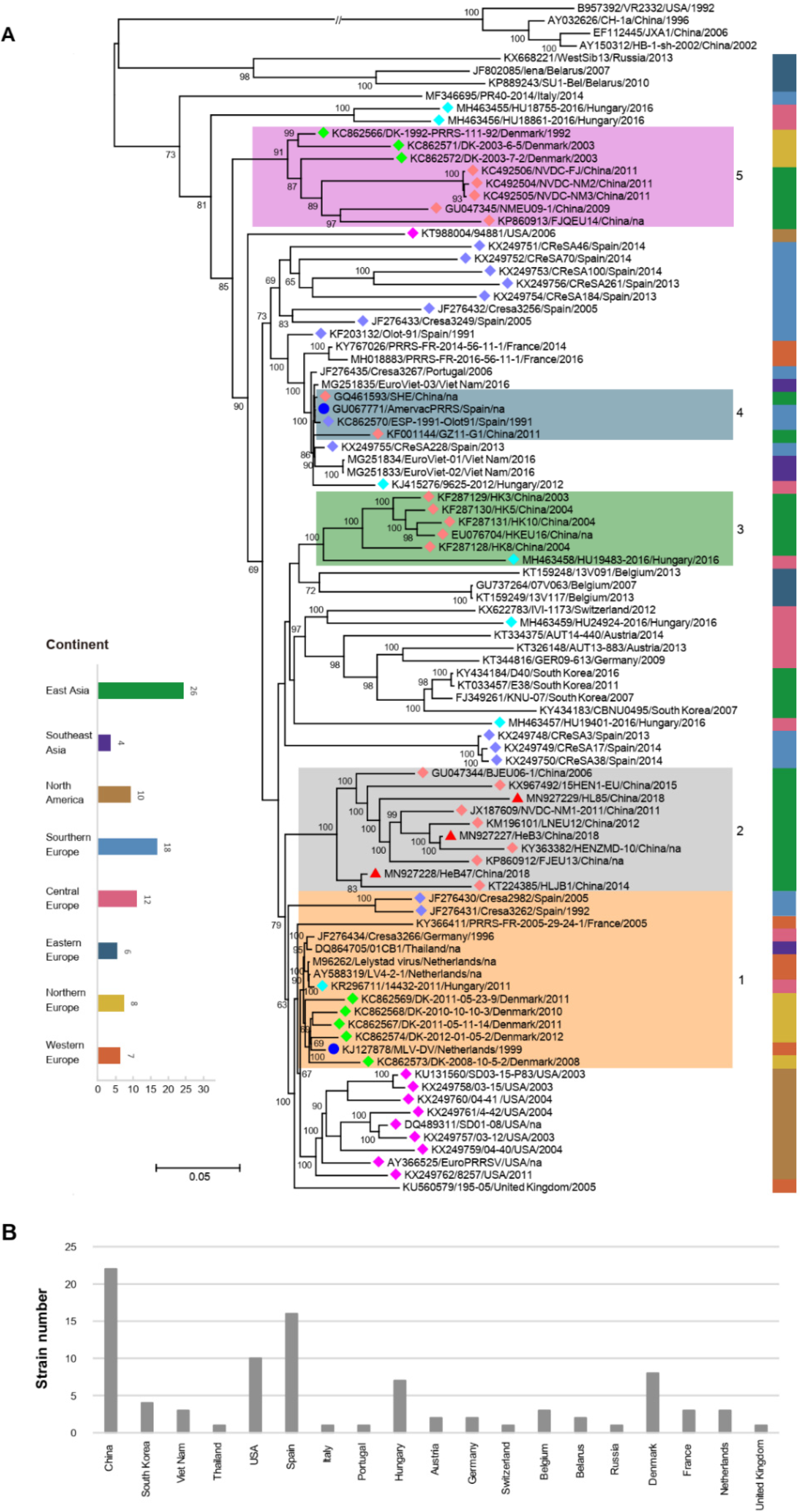
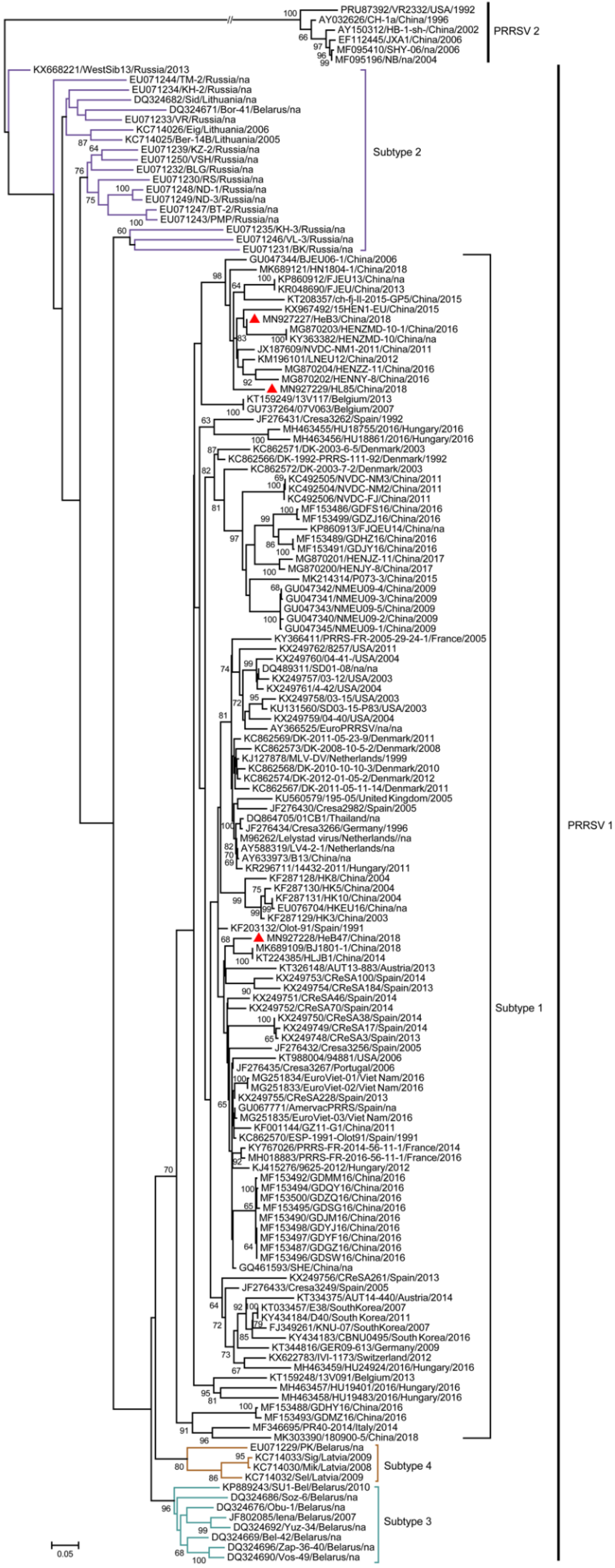
3.6. Phylogenetic Analysis of Three Samples Based on the Complete Genome Sequences
3.7. Phylogenetic Analysis of Three Samples Based on the ORF5 Genome Sequences
3.8. Establishment of RT-qPCR for PRRSV-1 Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neumann, E.J.; Kliebenstein, J.B.; Johnson, C.D.; Mabry, J.W.; Bush, E.J.; Seitzinger, A.H.; Green, A.L.; Zimmerman, J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the united states. J. Am. Vet. Med. Assoc. 2005, 227, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, K.; Yu, X.; Zhao, T.; Feng, Y.; Cao, Z.; Wang, C.; Hu, Y.; Chen, X.; Hu, D.; Tian, X.; et al. Emergence of fatal prrsv variants: Unparalleled outbreaks of atypical prrs in china and molecular dissection of the unique hallmark. PLoS ONE 2007, 2, e526. [Google Scholar] [CrossRef]
- Collins, J.E.; Benfield, D.A.; Christianson, W.T.; Harris, L.; Hennings, J.C.; Shaw, D.P.; Goyal, S.M.; McCullough, S.; Morrison, R.B.; Joo, H.S. Isolation of swine infertility and respiratory syndrome virus (isolate atcc vr-2332) in north america and experimental reproduction of the disease in gnotobiotic pigs. J. Vet. Diagn. Investig. 1992, 4, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Meulenberg, J.J. The molecular biology of arteriviruses. J. Gen. Virol. 1998, 79, 961–979. [Google Scholar] [CrossRef]
- Johnson, C.R.; Griggs, T.F.; Gnanandarajah, J.; Murtaugh, M.P. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative orf5 present in all arteriviruses. J. Gen. Virol. 2011, 92, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Treffers, E.E.; Li, Y.; Tas, A.; Sun, Z.; van der Meer, Y.; de Ru, A.H.; van Veelen, P.A.; Atkins, J.F.; Snijder, E.J.; et al. Efficient -2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. USA 2012, 109, E2920–E2928. [Google Scholar] [CrossRef] [Green Version]
- Meulenberg, J.J.M.; de Meijer, E.J.; Moormann, R.J.M. Subgenomic rnas of lelystad virus contain a conserved leader-body junction sequence. J. Gen. Virol. 1993, 74, 1697–1701. [Google Scholar] [CrossRef]
- Firth, A.E.; Zevenhoven-Dobbe, J.C.; Wills, N.M.; Go, Y.Y.; Balasuriya, U.B.R.; Atkins, J.F.; Snijder, E.J.; Posthuma, C.C. Discovery of a small arterivirus gene that overlaps the gp5 coding sequence and is important for virus production. J. Gen. Virol. 2011, 92, 1097–1106. [Google Scholar] [CrossRef]
- Das, P.B.; Dinh, P.X.; Ansari, I.H.; de Lima, M.; Osorio, F.A.; Pattnaik, A.K. The minor envelope glycoproteins gp2a and gp4 of porcine reproductive and respiratory syndrome virus interact with the receptor cd163. J. Virol. 2010, 84, 1731–1740. [Google Scholar] [CrossRef] [Green Version]
- Wissink, E.H.; Kroese, M.V.; van Wijk, H.A.; Rijsewijk, F.A.; Meulenberg, J.J.; Rottier, P.J. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J. Virol. 2005, 79, 12495–12506. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, J.H.; Lauck, M.; Bailey, A.L.; Shchetinin, A.M.; Vishnevskaya, T.V.; Bào, Y.; Ng, T.F.F.; LeBreton, M.; Schneider, B.S.; Gillis, A.; et al. Reorganization and expansion of the nidoviral family arteriviridae. Arch. Virol. 2016, 161, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Stadejek, T.; Stankevicius, A.; Murtaugh, M.P.; Oleksiewicz, M.B. Molecular evolution of prrsv in europe: Current state of play. Vet. Microbiol. 2013, 165, 21–28. [Google Scholar] [CrossRef]
- Allende, R.; Lewis, T.L.; Lu, Z.; Rock, D.L.; Kutish, G.F.; Ali, A.; Doster, A.R.; Osorio, F.A. North american and european porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J. Gen. Virol. 1999, 80, 307–315. [Google Scholar] [CrossRef]
- Dea, S.; Gagnon, C.A.; Mardassi, H.; Pirzadeh, B.; Rogan, D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (prrs) virus: Comparison of the north american and european isolates. Arch. Virol. 2000, 145, 659–688. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Schneider, P.; Zhang, W.P.; Faaberg, K.S.; Nelson, E.A.; Rowland, R.R. Diversity and evolution of a newly emerged north american type 1 porcine arterivirus: Analysis of isolates collected between 1999 and 2004. Arch. Virol. 2007, 152, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Frossard, J.P.; Hughes, G.J.; Westcott, D.G.; Naidu, B.; Williamson, S.; Woodger, N.G.; Steinbach, F.; Drew, T.W. Porcine reproductive and respiratory syndrome virus: Genetic diversity of recent british isolates. Vet. Microbiol. 2013, 162, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Liu, Q.; Qiao, M.; Deng, X.; Chen, X.; Sun, M. Whole genome characterization of a novel porcine reproductive and respiratory syndrome virus 1 isolate: Genetic evidence for recombination between amervac vaccine and circulating strains in mainland china. Infect. Genet. Evol. 2017, 54, 308–313. [Google Scholar] [CrossRef]
- Eclercy, J.; Renson, P.; Lebret, A.; Hirchaud, E.; Normand, V.; Andraud, M.; Paboeuf, F.; Blanchard, Y.; Rose, N.; Bourry, O. A field recombinant strain derived from two type 1 porcine reproductive and respiratory syndrome virus (prrsv-1) modified live vaccines shows increased viremia and transmission in spf pigs. Viruses 2019, 11, 296. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Cao, Z.; Yu, X.; Deng, X.; Zhao, T.; Wang, L.; Liu, Q.; Li, X.; Tian, K. Emergence of novel european genotype porcine reproductive and respiratory syndrome virus in mainland china. J. Gen. Virol. 2011, 92, 880–892. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Yang, C.; Zheng, Y.; Gauger, P.C.; Anderson, T.; Harmon, K.M.; Zhang, J.; Yoon, K.J.; Main, R.G.; et al. The emergence of novel sparrow deltacoronaviruses in the united states more closely related to porcine deltacoronaviruses than sparrow deltacoronavirus hku17. Emerg. Microbes Infect. 2018, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in india, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. Raxml-ng: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lam, T.T.; Hon, C.C.; Hui, R.K.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C. Molecular epidemiology of prrsv: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Q.; Hu, D.; Zhang, Q.; Han, T.; Ma, Y.; Gu, X.; Zhai, X.; Tian, K. Complete genomic characterization and genetic diversity of four european genotype porcine reproductive and respiratory syndrome virus isolates from china in 2011. Virus Genes 2015, 51, 375–384. [Google Scholar] [CrossRef]
- Liu, J.K.; Wei, C.H.; Dai, A.L.; Fan, K.W.; Yang, B.H.; Huang, C.F.; Li, X.H.; Yang, X.Y.; Luo, M.L. Complete genomic characterization of two european-genotype porcine reproductive and respiratory syndrome virus isolates in fujian province of china. Arch. Virol. 2017, 162, 823–833. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate rna virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Oleksiewicz, M.B.; Botner, A.; Toft, P.; Normann, P.; Storgaard, T. Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: The nsp2 fragment of the replicase polyprotein contains a cluster of b-cell epitopes. J. Virol. 2001, 75, 3277–3290. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, X.; Zhou, R.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Genomic characterization and pathogenicity of a strain of type 1 porcine reproductive and respiratory syndrome virus. Virus Res. 2016, 225, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ropp, S.L.; Wees, C.E.; Fang, Y.; Nelson, E.A.; Rossow, K.D.; Bien, M.; Arndt, B.; Preszler, S.; Steen, P.; Christopher-Hennings, J.; et al. Characterization of emerging european-like porcine reproductive and respiratory syndrome virus isolates in the united states. J. Virol. 2004, 78, 3684–3703. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, J.; Zeng, J.; Yin, S.; Li, Y.; Zheng, L.; Guo, X.; Ge, X.; Yang, H. The 30-amino-acid deletion in the nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in china is not related to its virulence. J. Virol. 2009, 83, 5156–5167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Wei, Z.; Zevenhoven-Dobbe, J.C.; Liu, R.; Tong, G.; Snijder, E.J.; Yuan, S. Arterivirus minor envelope proteins are a major determinant of viral tropism in cell culture. J. Virol. 2012, 86, 3701–3712. [Google Scholar] [CrossRef] [Green Version]
- Marcuello, C.; de Miguel, R.; Lostao, A. Molecular recognition of proteins through quantitative force maps at single molecule level. Biomolecules 2022, 12, 594. [Google Scholar] [CrossRef] [PubMed]
- Bian, T.; Sun, Y.; Hao, M.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. A recombinant type 2 porcine reproductive and respiratory syndrome virus between nadc30-like and a mlv-like: Genetic characterization and pathogenicity for piglets. Infect. Genet. Evol. 2017, 54, 279–286. [Google Scholar] [CrossRef]
- Zhao, H.; Han, Q.; Zhang, L.; Zhang, Z.; Wu, Y.; Shen, H.; Jiang, P. Emergence of mosaic recombinant strains potentially associated with vaccine jxa1-r and predominant circulating strains of porcine reproductive and respiratory syndrome virus in different provinces of china. Virol. J. 2017, 14, 67. [Google Scholar] [CrossRef]
- Dortmans, J.; Buter, G.J.; Dijkman, R.; Houben, M.; Duinhof, T.F. Molecular characterization of type 1 porcine reproductive and respiratory syndrome viruses (prrsv) isolated in the netherlands from 2014 to 2016. PLoS ONE 2019, 14, e0218481. [Google Scholar] [CrossRef]
- Martin-Valls, G.E.; Kvisgaard, L.K.; Tello, M.; Darwich, L.; Cortey, M.; Burgara-Estrella, A.J.; Hernandez, J.; Larsen, L.E.; Mateu, E. Analysis of orf5 and full-length genome sequences of porcine reproductive and respiratory syndrome virus isolates of genotypes 1 and 2 retrieved worldwide provides evidence that recombination is a common phenomenon and may produce mosaic isolates. J. Virol. 2014, 88, 3170–3181. [Google Scholar] [CrossRef] [Green Version]
- Kvisgaard, L.K.; Kristensen, C.S.; Ryt-Hansen, P.; Pedersen, K.; Stadejek, T.; Trebbien, R.; Andresen, L.O.; Larsen, L.E. A recombination between two type 1 porcine reproductive and respiratory syndrome virus (prrsv-1) vaccine strains has caused severe outbreaks in danish pigs. Transbound. Emerg. Dis. 2020, 67, 1786–1796. [Google Scholar] [CrossRef] [Green Version]
- Renson, P.; Touzain, F.; Lebret, A.; Le Dimna, M.; Quenault, H.; Normand, V.; Claude, J.B.; Pez, F.; Rose, N.; Blanchard, Y.; et al. Complete genome sequence of a recombinant porcine reproductive and respiratory syndrome virus strain from two genotype 1 modified live virus vaccine strains. Genome Announc. 2017, 5, e00454-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhou, X.; Lunney, J.K.; Lawson, S.; Sun, Z.; Brown, E.; Christopher-Hennings, J.; Knudsen, D.; Nelson, E.; Fang, Y. Immunodominant epitopes in nsp2 of porcine reproductive and respiratory syndrome virus are dispensable for replication, but play an important role in modulation of the host immune response. J. Gen. Virol. 2010, 91, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kaiser, T.J.; Horlen, K.; Keith, M.L.; Taylor, L.P.; Jolie, R.; Calvert, J.G.; Rowland, R.R. Insertion and deletion in a non-essential region of the nonstructural protein 2 (nsp2) of porcine reproductive and respiratory syndrome (prrs) virus: Effects on virulence and immunogenicity. Virus Genes 2009, 38, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.Q.; Guo, X.; Yang, H.C. Genomic characterization of two chinese isolates of porcine respiratory and reproductive syndrome virus. Arch. Virol. 2004, 149, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Li, Z.; Xia, M.; Li, X.; Wang, F.; Wang, W.; Zhang, X.; Wu, H. Mutations in the genome of the highly pathogenic porcine reproductive and respiratory syndrome virus potentially related to attenuation. Vet. Microbiol. 2012, 157, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Montagnaro, S.; Sasso, S.; De Martino, L.; Longo, M.; Iovane, V.; Ghiurmino, G.; Pisanelli, G.; Nava, D.; Baldi, L.; Pagnini, U. Prevalence of antibodies to selected viral and bacterial pathogens in wild boar (sus scrofa) in campania region, italy. J. Wildl. Dis. 2010, 46, 316–319. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.; Charbonneau, G.; Carman, S.; Hamel, A.; Nayar, G.; Friendship, R.; Eernisse, K.; Swenson, S. Lelystad-like strain of porcine reproductive and respiratory syndrome virus (prrsv) identified in canadian swine. Can. Vet. J. 2000, 41, 493–494. [Google Scholar]
- Lee, C.; Kim, H.; Kang, B.; Yeom, M.; Han, S.; Moon, H.; Park, S.; Kim, H.; Song, D.; Park, B. Prevalence and phylogenetic analysis of the isolated type i porcine reproductive and respiratory syndrome virus from 2007 to 2008 in korea. Virus Genes 2010, 40, 225–230. [Google Scholar] [CrossRef]
- Greiser-Wilke, I.; Fiebig, K.; Drexler, C.; grosse Beilage, E. Genetic diversity of porcine reproductive and respiratory syndrome virus (prrsv) in selected herds in a pig-dense region of north-western germany. Vet. Microbiol. 2010, 143, 213–223. [Google Scholar] [CrossRef]
- Prieto, C.; Vázquez, A.; Núñez, J.I.; Alvarez, E.; Simarro, I.; Castro, J.M. Influence of time on the genetic heterogeneity of spanish porcine reproductive and respiratory syndrome virus isolates. Vet. J. 2009, 180, 363–370. [Google Scholar] [CrossRef]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical genome mapping: A promising new tool to assess genomic complexity in chronic lymphocytic leukemia (cll). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Li, S.T.; Yao, W.Y.; Xie, C.D.; Chen, Z.; Zeng, Z.J.; Wang, D.; Xu, K.; Shen, Z.J.; Mu, Y.; et al. The meishan pig genome reveals structural variation-mediated gene expression and phenotypic divergence underlying asian pig domestication. Mol. Ecol. Resour. 2021, 21, 2077–2092. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % of Identity to Lelystad Virus | |||
|---|---|---|---|
| HeB3 | HeB47 | HL85 | |
| Complete genome (nt) | 91.3 | 93.8 | 88.4 |
| pp1a (aa) | 89.8 | 92.3 | 78.7 |
| pp1ab (aa) | 97.5 | 98.4 | 96.6 |
| GP2 (aa) | 94 | 94.8 | 90.4 |
| GP3 (aa) | 90.6 | 92.9 | 78.9 |
| GP4 (aa) | 91.2 | 90.7 | 89.5 |
| GP5 (aa) | 89.6 | 91.6 | 87.6 |
| M (aa) | 96.5 | 98.3 | 96.5 |
| N (aa) | 95.3 | 96.1 | 91.5 |
| Standard Plasmid (Copies/μL) | Reproducibility (Inter-Assay) | Repeatability (Intra-Assay) | ||
|---|---|---|---|---|
| Mean ± SD | CV | Mean ± SD | CV | |
| High (2.5 × 106) | 19.01 ± 0.03 | 0.16 | 19.15 ± 0.11 | 0.49 |
| Medium (2.5 × 104) | 25.59 ± 0.16 | 0.63 | 25.67 ± 0.06 | 0.23 |
| Low (2.5 × 102) | 32.51 ± 0.04 | 0.12 | 32.35 ± 0.09 | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.; Liu, L.; Tian, X.; Chen, L.; Huang, X.; Sun, Y.; Yan, Y.; Tian, Z.; Cai, X.; Liu, D.; et al. Genomic Analysis of Porcine Reproductive and Respiratory Syndrome Virus 1 Revealed Extensive Recombination and Potential Introduction Events in China. Vet. Sci. 2022, 9, 450. https://doi.org/10.3390/vetsci9090450
Yu F, Liu L, Tian X, Chen L, Huang X, Sun Y, Yan Y, Tian Z, Cai X, Liu D, et al. Genomic Analysis of Porcine Reproductive and Respiratory Syndrome Virus 1 Revealed Extensive Recombination and Potential Introduction Events in China. Veterinary Sciences. 2022; 9(9):450. https://doi.org/10.3390/vetsci9090450
Chicago/Turabian StyleYu, Fang, Liqiang Liu, Xiaoxiao Tian, Ligong Chen, Xinyi Huang, Yue Sun, Yi Yan, Zhijun Tian, Xuehui Cai, Di Liu, and et al. 2022. "Genomic Analysis of Porcine Reproductive and Respiratory Syndrome Virus 1 Revealed Extensive Recombination and Potential Introduction Events in China" Veterinary Sciences 9, no. 9: 450. https://doi.org/10.3390/vetsci9090450
APA StyleYu, F., Liu, L., Tian, X., Chen, L., Huang, X., Sun, Y., Yan, Y., Tian, Z., Cai, X., Liu, D., & An, T. (2022). Genomic Analysis of Porcine Reproductive and Respiratory Syndrome Virus 1 Revealed Extensive Recombination and Potential Introduction Events in China. Veterinary Sciences, 9(9), 450. https://doi.org/10.3390/vetsci9090450