1. Introduction
Brucellosis is a zoonotic disease that causes abortion and infertility in animals and undulant fever in humans [
1]. It is distributed worldwide and endemic in developing countries. It is caused by the genus
Brucella [
2] and belongs to the order alpha-Proteobacteria, which consists of mostly intra-cellular bacteria that are recognised as pathogenic in a number of mammalian hosts [
3]. Twelve
Brucella species have been characterised: six classical species, which includes
Brucella suis,
B. melitensis,
B. abortus,
B. ovis,
B. neotomae, and
B. canis; an additional six novel species, which includes
B. ceti from cetaceans,
B. pinnipedialis from seals [
4],
B. microti from wild rodents,
B. inopinata from breast implant infection in humans [
5],
B. vulpis from red foxes [
6], and
B. papionis from baboons [
7]. These are classified based on distinction between host preference and pathogenicity [
8]. The smooth strains (
B. suis,
B. melitensis, and
B. abortus) are all zoonotic, and of the rough strains (
B. ovis and
B. canis), only
B. canis is zoonotic [
9].
Brucellosis is a controlled and notifiable disease in humans and animals across sub-Saharan Africa [
10]. Clinical signs vary in animals depending on the host species [
11]. These include spontaneous abortion, weak offspring, pyrexia, hygromas and mastitis [
12], orchitis, epididymitis, ampullitis, and seminal vesiculitis [
13]. However, these clinical presentations are non-specific, making diagnosis difficult. In addition, latent asymptomatic heifers introduced into a naïve herd cause abortion storms. The placenta, foetal fluid, and aborted foetus serve as major sources of infection to other animals [
14].
The diagnosis of the disease is by routine serological tests and bacterial culture isolation, which is the “gold standard” in brucellosis diagnosis [
15]. However, these tests have inadequate sensitivity and specificity, are time-consuming, and have the potential of causing infection in laboratory personnel. To overcome these challenges, several conventional and real-time PCR assays utilising different primers derived from different polymorphic regions in the
Brucella genome have been developed [
16,
17]. While these assays generally work well, immunological cross reactions with other closely related species such as
Ochrobactrum anthropi have been reported in conventional PCR assays [
18], resulting in reduced assay specificity [
19]. Some of these assays are only able to differentiate a limited number of species [
20]. There is a need to develop accurate and rapid diagnostic assays for high-throughput detection of
Brucella, in order to support appropriate control measures and contribute to the eradication of the disease in a country.
The 16S-23S ribosomal deoxyribonucleic acid (rDNA) internal transcribed spacer region (ITS) PCR has proved to be a promising diagnostic tool. The 16S-23S ITS has also been used in the diagnosis of brucellosis in humans [
21]. It has been proven to be rapid, specific, and more sensitive for the diagnosis of brucellosis in both animals and humans because the rDNA exists in multiple copies [
22]. The ITS region is highly conserved, with 100% sequence homology among
Brucella spp. [
23].
The aim of this study was to develop a rapid genus-specific qPCR assay to detect Brucella species targeting the 16S-23S rDNA ITS region.
2. Materials and Methods
2.1. Assay Design
Using
B. abortus (GenBank accession number of X95889) as a template, nucleic acid sequences of the 16S-23S rDNA ITS region of
Brucella spp. were retrieved from GenBank [
24], using the nucleotide Basic Local Alignment Search Tool (BLAST
®, National Center for Biotechnology Information, Bethesda, MD, USA) [
24,
25], and the highly similar sequences programme (megablast, National Center for Biotechnology Information, Bethesda, MD, USA). An
O. intermedium sequence (AJ867325) was used as a template, together with the discontiguous megablast programme, to obtain sequences of other bacteria with close homology to
Brucella.
The retrieved genetic sequences were aligned online using a multiple sequence alignment programme (MAFFT version 7) [
26]. Data analysis in molecular biology and evolution software (DAMBE) [
27] was used to identify and remove identical sequences and sequences were edited with BioEdit [
28]. A TaqMan
® minor groove binder (MGB) assay was designed using Primer Express v3.0 using default primer/probe design parameters: melting temperature (Tm) of 58–60 °C and 68–70 °C respectively, a GC% content of 30–80%, the last five nucleotides of the 3′ end of the primer do not contain more than two ‘G’ and ‘C’ residues, and the 5′ end of the probe does not contain a ‘G’ residue (ThermoFisher Scientific, Waltham, MA, USA). The amplicon length was 72 bp.
Field strains of
Brucella spp. obtained from the Department of Veterinary Tropical Disease (DVTD) diagnostics laboratory, University of Pretoria (UP) and closely related non-
Brucella species, such as
Ochrobactrum anthropi were used for this study (
Table 1). All bacterial isolates were sub-cultured on blood agar at 37 °C for 72 h for
Brucella spp. and 24 h for other bacteria [
22].
Chlamydia abortus was cultured on a liquid yeast based medium, serum casein glucose yeast extract medium recommended by the supplier of the control strain. The host used was
Acanthamoeba castellani. Two millilitres (mL) of medium and 20–50 microlitres (µL) of
Acanthamoeba castellani in a plastic 5 mL screw top tube was used. After three days when a white precipitate at the bottom of the tube was seen, 50 µL of
Chlamydia abortus was added and reincubated in normal air at 30 °C for seven days.
Cultures were obtained from the DVTD diagnostic laboratory. Colonies were harvested and stored in 10% glycerol and stored at −80 °C for further use. Then, these were thawed, and sub-cultures were prepared. These were streaked and inoculated on Selecta-MEDIA Blood Tryptose agar +5% sheep blood agar (ThermoFisher Scientific, Waltham, MA, USA). The Brucella strains were cultured in an HF151UV CO2 incubator (Heal Force, Shangai, China) for 72 h. Bacterial colonies on the plates were scraped off using 10 µL sterile inoculating loops and transferred to 1.5 mL Eppendorf tubes containing 200 µL phosphate-buffered saline (PBS) (Sigma-Aldrich, St Louis, MO, USA). Bacteria were quantified using a TC20™ automated cell counter (Bio-Rad Laboratories, Singapore).
2.2. Control Samples
A negative blood control sample was collected from a cow with no history of brucellosis. A complete blood count and microscopic examination for haemoparasites was performed on the sample by the Clinical Pathology Laboratory, Onderstepoort Veterinary Teaching Hospital, UP. A negative fresh milk (bulk tank milk) control sample was collected from a farm with no history of brucellosis, located at Irene, Gauteng, South Africa. Negative tissue control samples from the abattoir were collected from bovine, caprine, and ovine. All samples (blood, milk, and tissue) tested negative for Brucella spp. by the ITS qPCR assay.
Brucella colonies cultured on blood agar medium were scraped off the culture plates using a sterile inoculating loop and harvested into 1 mL of PBS (Sigma-Aldrich, St Louis, MO, USA). This was the stock solution used for all experiments throughout the study. The number of cells/mL of stock solution (Brucella abortus bv. 1 (B01988-18 strain) with a bacterial cell count of 3.19 × 107 cells/mL) was determined using a TC20™ automated cell counter (Bio-Rad Laboratories, Hercules, CA, USA). Blood and milk samples (180 µL) were spiked with 20 µL of a stock B. abortus solution and 20–30 milligrams (mg) of Brucella positive foetal aborted tissue (confirmed by culture isolation) were tested by the ITS qPCR assay and used as positive controls.
2.3. Nucleic Acid Purification
Nucleic acid was purified using the KingFisher™ Duo Prime purification system and the MagMAX CORE™ Nucleic Acid Purification Kit (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Nucleic acid from blood and milk samples (200 µL) was purified using the simple workflow and eluted in 90 µL of elution buffer before storage of the DNA at −20 °C. The simple and digestion methods were compared in purifying DNA from aborted foetal tissues. The latter was determined to be superior, as this method produced lower cycle threshold results with higher fluorescence on qPCR. Proteinase K (PK) solution, composed of (PK Buffer and MagMax CORE Proteinase K) was added to approximately 20–30 mg of tissue and incubated for 2 h at 55 °C. The supernatant was processed as recommended by the manufacturer. The complex workflow was used to purify DNA from 300 µL foetal abomasal fluid.
VetMAX™ Xeno™ Internal Positive Control (IPC) DNA was added in all workflows.
2.4. Real-Time PCR
The VetMAX™-Plus qPCR Master Mix (ThermoFisher Scientific, Waltham, MA, USA) was used for DNA amplification. The PCR amplification mixture comprised 12.5 µL 2× qPCR Master Mix, 400 nM (final concentration) forward/reverse primers, 150 nM (final concentration) probe, 1 µL VetMAX™ Xeno™ Internal Positive Control-VIC™ Assay (ThermoFisher Scientific, Waltham, MA, USA), 2 µL template DNA, and nuclease-free water to make up a total volume of 25 µL per reaction.
Detection and amplification of purified DNA by qPCR was performed on the StepOnePlus™ Real-Time PCR System running StepOne Software v2.3 system (ThermoFisher Scientific, Waltham, MA, USA). The cycling conditions recommended by the manufacturer of the PCR reagents were used.
2.5. Assay Optimisation
The primer and probe concentration of the genus-specific qPCR assay was optimised by testing primer/probe concentrations in triplicates for B. melitensis (BMR 18001) and B. abortus (B01988-18). For primer optimisation, a combination of four primer concentrations in the PCR reaction (800, 400, 200, and 100 nM) of both forward and reverse primers were tested, with a constant probe concentration in the PCR reaction of 250 nM. Then, the optimised primer concentration determined was used for probe optimisation by varying probe concentrations in the PCR reaction (250, 200, 150, 100, and 50 nM). The combination of primer and TaqMan® probe concentration that yielded optimal assay performance (low cycle threshold (CT), efficient (steep) amplification slope, and low primer/probe concentration) was chosen for further experiments.
2.6. Analytical Validation
The analytical performance of the assay was determined by following the Stage 1 validation pathway in Chapter 1.1.2 of the OIE Manual of Diagnostic Tests and Vaccines for Aquatic Animals of the World Organisation for Animal Health [
29].
The intended purpose of the assay is to (i) contribute to the eradication or elimination of brucellosis from defined populations, (ii) confirm the diagnosis of suspect or clinical cases and (iii) estimate the prevalence of infection or exposure to facilitate risk analysis (surveys, herd health status, disease control measures) in samples submitted for veterinary diagnostics.
2.6.1. Efficiency
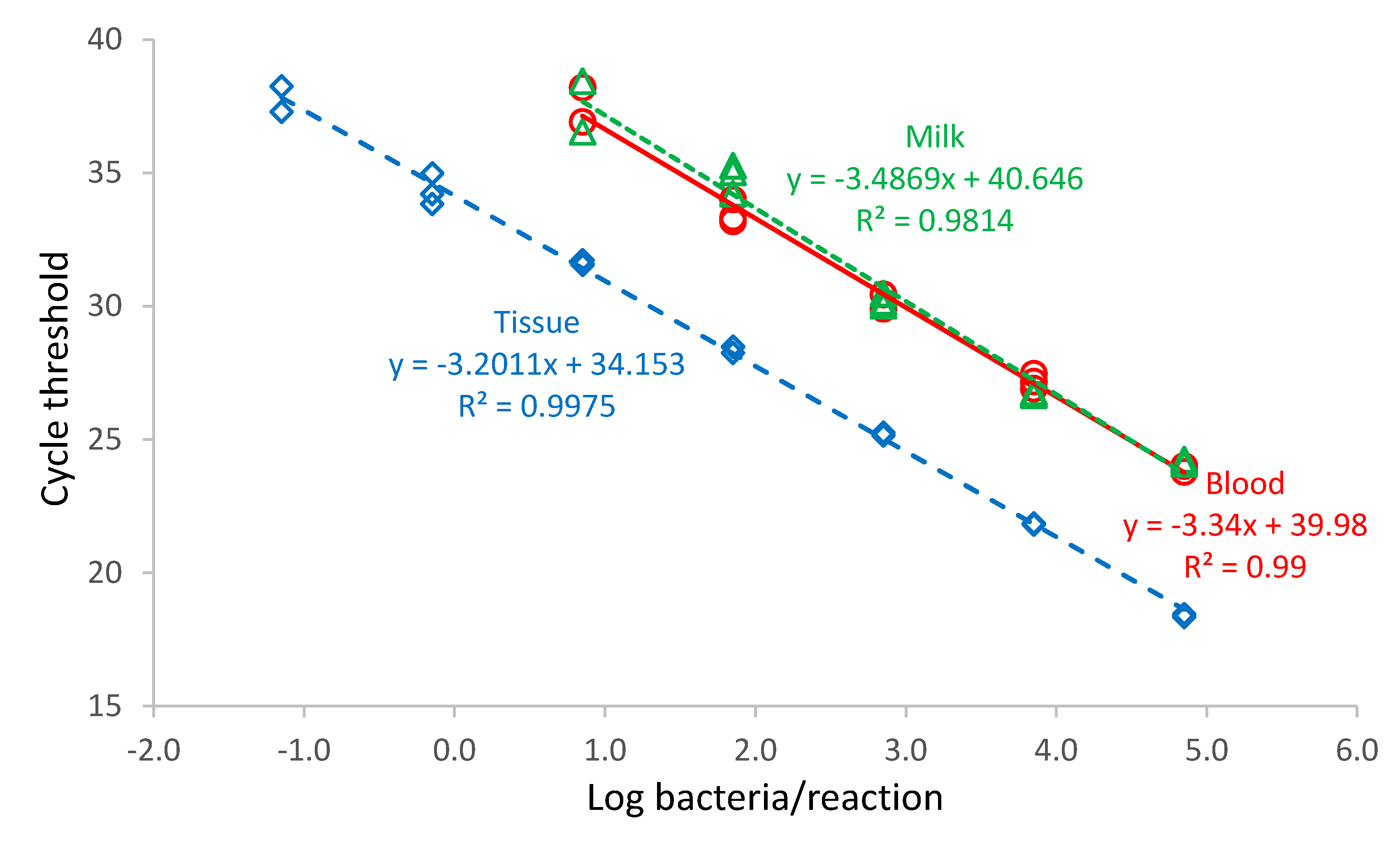
Brucella abortus bv. 1 (B01988-18 strain) with a bacterial cell count of 3.19 × 107 cells/mL was diluted with PBS in a ten-fold dilution series from 10−8–100 Blood, milk, and digested tissue sample matrix (180 µL) samples were spiked with 20 µL of the B. abortus dilution. DNA was purified from each dilution, and the PCR was performed in triplicate.
Standard curves for each run and dilution series were plotted showing CT-values versus log bacteria/PCR reaction. Linear regressions of the data were performed using Microsoft Excel and the slopes of the regression lines of the standard curve used to calculate the efficiency of the PCR assay (%) = (10−1/slope − 1) × 100.
2.6.2. Analytical Sensitivity
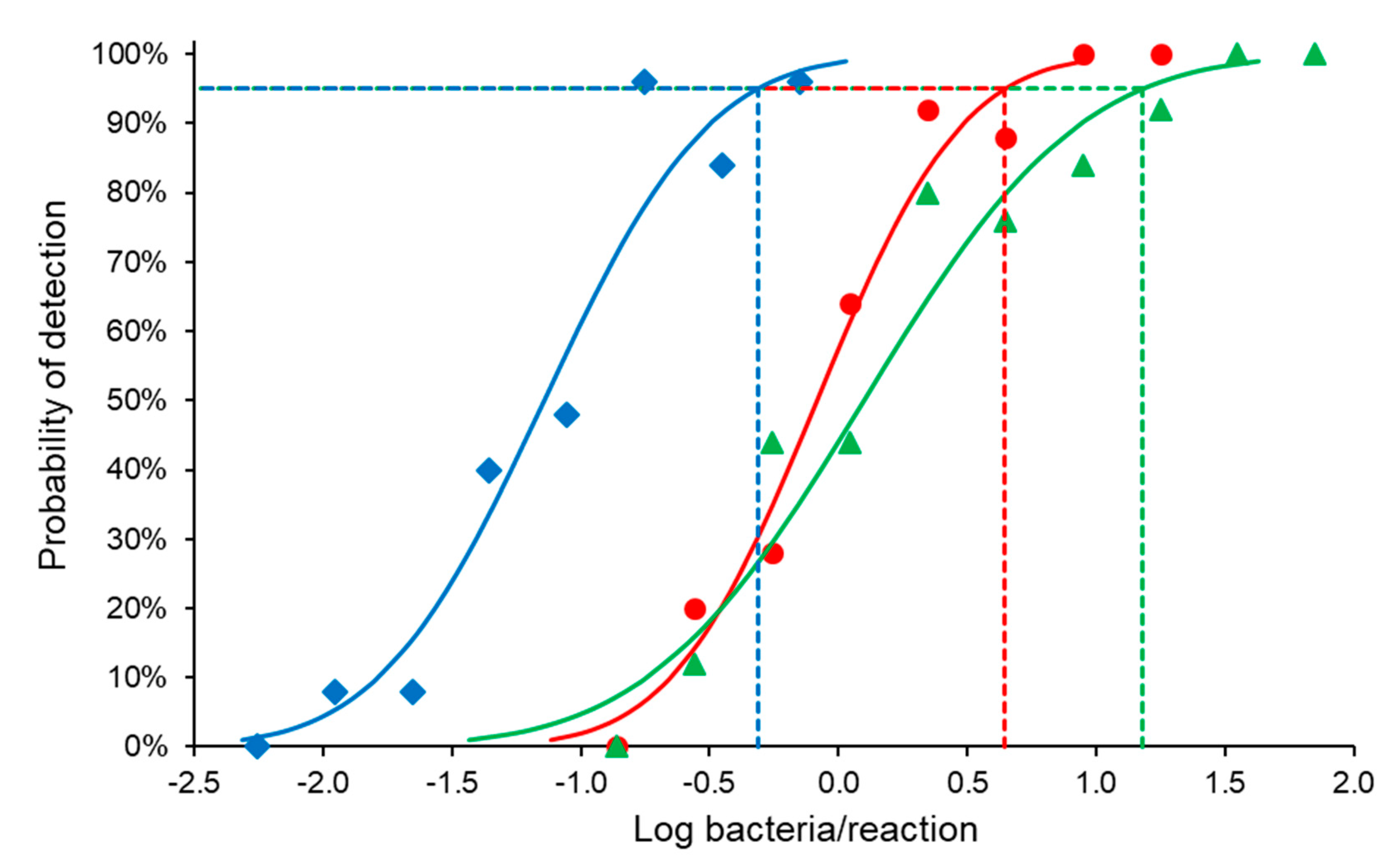
The last dilution that produced a positive result in all replicates, obtained from the efficiency analysis (70.89 bacteria/PCR reaction), was used to prepare a two-fold dilution series of B. abortus bv. 1 (B01988-18 strain) with PBS to cover the non-linear range of the assay. Blood, milk, and tissue sample matrix were spiked with B. abortus. DNA from each dilution was purified five times, and each purified DNA sample was tested by PCR five times (each dilution was tested 25 times in total) for all matrixes.
SPSS Statistics 25 software (IBM, New York, NY, USA) was used to perform a probit analysis to determine the 95% limit of detection (LOD).
There are three genome copies of the 16S-23S ITS region per
Brucella sp. bacterium [
21]. Results were expressed in bacterial copies, where one bacterium is equivalent to three genome copies.
2.6.3. Analytical Specificity
The ITS qPCR assay was tested against a panel of Gram-positive and Gram-negative bacteria, as listed in
Table 1. Bacterial strains were cultured, and a loop full of colonies was harvested and mixed well in 1 mL of PBS for all the strains in Eppendorf tubes. The mixtures were diluted 1:10 in PBS before the DNA was purified and tested by PCR, as described previously.
The O. intermedium bacterium was not available in our laboratory as a bacterial culture, and the DNA sequence of O. intermedium that encompassed the target region of the PCR assay was synthesised by IDT (Integrated DNA Technologies, Coralville, IA, USA).
2.6.4. Repeatability
Data obtained from the sensitivity results were used to determine the inter-run and intra-run repeatability of the assay, using Microsoft Excel.
2.7. Diagnostic Performance on Field Samples
2.7.1. Sample Preparation
A blind study of random positive and negative samples was obtained from the Bacteriology laboratory of the Department of Veterinary Tropical Diseases. A total of 56 aborted foetal tissue samples were used to evaluate the diagnostic performance of the assay. The samples comprised 24 foetal abomasal fluid samples and 32 tissue samples (which included aborted foetal liver, lung and placenta from bovine, caprine, and ovine). DNA from these samples were purified and tested using the ITS PCR and the BCSP31 PCR [
16] assays in parallel.
2.7.2. Real-Time PCR
The developed ITS PCR assay was compared to the BCSP31 PCR assay for diagnostic validation because the BCSP31 gene target is one of the most commonly used targets used for the detection of
Brucella spp. in animals [
30] and is routinely used by OIE reference laboratories. The BCSP31 PCR can be used in a real-time PCR format, and the primers and probe for the
Brucella spp. described by Probert et al. [
16] was used in our study. This assay was used on a different platform (StepOne real-time PCR machine, Applied Biosystems™, Foster City, CA, USA) and the VetMAX™-Plus qPCR Master Mix (as described in 2.4, but with 300 nM forward and reverse primer concentrations and 100 nM probe concentration in the final reaction.
2.7.3. Diagnostic Sensitivity and Specificity Statistical Analysis
Diagnostic sensitivity and specificity of the PCR assay were estimated in the absence of a gold standard assay, by using a three-test one-population Bayesian latent class model that allowed for conditional dependence between two of the tests and independence of the third test [
31].
Modes were obtained from published references [
21,
22,
32,
33]. For multiple references, the average of the modes was used (
Table 2). For the sensitivity of the 16S-23S rDNA ITS PCR assay, the references reported on the diagnostic sensitivity in blood samples. For the diagnostic sensitivity in aborted material, the prior for this parameter was adjusted upwards. No reference could be obtained for “prevalence”, the proportion of tested aborted material positive for
Brucella spp. submitted to a diagnostic laboratory in South Africa and expert opinion (Dr. A. Jonker, DVTD, UP) was obtained.
The model was run in OpenBUGS, version 3.2.3 rev 1012, a programme for Bayesian analysis of complex statistical models using Markov chain Monte Carlo techniques [
34,
35]. Two chains were used, and initial values were generated by forward sampling from the prior distribution for each parameter. The first 10,000 iterations were discarded, and the next 50,000 iterations were used for posterior inferences. The first 10,000 iterations were discarded because it was regarded as “burn-in” to check convergence. The simulation was run for a further number of iterations to obtain samples that could be used for posterior inference. Model convergence was assessed by visual inspection of the trace plots.
2.8. Ethics Statement
Ethics approval was obtained from the University of Pretoria Animal Ethics Committee with the project identification code (V004-19) on 29 January 2019. Section 20 approval was granted according to Act 35 of 1984 by the Directorate of Animal Health, South Africa on 8 January 2019.
4. Discussion
We have described the development and analytical validation of a genus-specific TaqMan
® MGB qPCR assay to detect
Brucella species targeting the 16S-23S rDNA ITS region. Keid et al. [
22] developed a conventional ITS PCR to detect
Brucella in various samples, including whole blood, serum, semen, and vaginal swabs, but this study was carried out exclusively in dogs. Real-time PCR has an advantage over conventional PCR because it is more sensitive and is a quicker assay to perform. Kattar et al. [
21] developed an ITS qPCR assay to detect
Brucella in humans; however, the performance of the ITS qPCR assay in detecting
Brucella in animal specimens was not known. This study filled the gap by adding the advantage of detecting
Brucella species in easily accessible samples (milk, blood, and tissues) for early detection, which is a more sensitive qPCR ITS assay as compared to Kattar et al. (2007), and lastly, this assay has been optimised in animal samples.
Prior to design of the assay, an in silico analysis was carried out to determine conserved region of the 16S-23S rDNA ITS region of all Brucella species as well as closely related bacteria, using the BLAST® analysis on GenBank.
The BLAST
® tool showed in silico specificity of the assay. In the laboratory, specificity was determined by testing
Brucella species against a panel of closely related bacteria. The developed ITS PCR assay showed good specificity by amplifying DNA from all
Brucella species and not amplifying closely related bacterial species, such as
Ochrobactrum anthropi and
O. intermedium. Our results were in agreement with Keid et al. [
22]; their ITS-PCR was specific, since there was no amplification of
O. anthropi DNA; however, this was a conventional ITS PCR. The same result was obtained by Kattar et al. [
21], who used hybridisation probes specific to the ribosomal 16S–23S ITS region,
omp31 and
omp25. All three assays exhibited an analytical sensitivity and specificity of 100%.
The developed ITS assay efficiency was 99% in blood, 93% in milk, and 105% in tissues. An efficiency exceeding 100% was recorded in tissues. This may be a result of the digestion method used to purify DNA from tissues, which differed from the method used for liquid samples by an additional incubation step of the tissue sample in proteinase kinase before purification of the nucleic acid. The digestion of tissue and
Brucella, a difficult organism to lyse, with PK solution enhances lysing of the bacteria [
37]. This may have caused the higher efficiency value in tissues compared to blood and milk.
The 95% LOD of the developed ITS-PCR assay was 4.43, 15.18, and 0.49
Brucella organisms/PCR reaction in blood, milk, and tissues respectively of
B. abortus bv. 1 (B01988-18 strain), which demonstrated the sensitivity of the assay. This is of diagnostic importance, because latent asymptomatic animals serve as a source of infection to other animals (maintaining the disease), as well as humans [
12]. In humans, brucellosis can progress to sub-clinical or chronic stages if not detected early and treated promptly [
38].
The increased sensitivity of the assay in tissue samples may be a result of the different nucleic acid purification protocol used for tissue, compared to liquid samples. The tissue protocol included an additional proteinase kinase digestion step, which may have increased the intra-cellular release of the bacteria, allowing for the more sensitive detection of Brucella organisms. Investigation into the addition of a digestion step for liquid samples when purifying nucleic acid, to increase the sensitivity of detection of Brucella organisms is warranted.
The standard deviation of the inter-run and intra-run reproducibility was in the range of 0.1 to 1.7. The CV ranged from 1.7% to 4.7%. Minimal variation in results was demonstrated.
A blind test was carried out on 56 samples that were obtained from the bacteriology laboratory (DVTD, UP), to determine this assay’s potential as a screening tool in an abortion panel. Nucleic acid was purified and amplified by ITS and BCSP31 qPCR assays. Then, test results were compared to culture results. Samples that were positive on ITS but negative on both BCSP31 and culture were retested by bacterial culture. It was interesting to note that when these samples were re-cultured, there was growth of
Brucella colonies, which were initially negative on culture. This demonstrated that
Brucella cases may be “missed” with traditional diagnostic techniques. It also confirms that brucellosis is under-reported in the country, since bacterial culture isolation is the “gold standard” of identification. The initial negative results obtained from culture could be as a result of the use of selective media. Farrell’s selective media is used mostly for the isolation of the smooth
Brucella, and this media inhibits the growth of
B. ovis as well as some
B. melitensis and
B. abortus strains [
39]. It may be postulated that these tissue samples could be infected with any of these three
Brucella species and supports the assertion that the developed ITS assay can detect any species of
Brucella.
We compared the diagnostic sensitivity and specificity of the ITS and BCSP31 qPCR assays in aborted tissue samples. The two assays were performed on the same PCR platform to provide a direct comparison between the two assays, but the PCR platform used differed from the platform published for the BCSP31 assay. To the best of our knowledge, there is no publication on the validation of an ITS qPCR assay to detect
Brucella spp. in aborted tissue samples. Out of the 56 samples tested, 9 were positive with all three tests, 37 were negative with all tests, 7 were negative with both bacterial culture and the BCSP31 assay but positive on the ITS assay: this could be a result of small amounts of target DNA in these samples. One sample was positive with both the ITS and BCSP31 assays but negative on bacterial culture. This may indicate the presence of dead organisms in this tissue sample. Two samples were positive with both bacterial culture and the ITS assay but negative with the BCSP31 assay. This could be explained because of the low number of bacteria in this sample. The C
T values of the ITS-PCR for these samples were 37.19 and 36.42, which indicated that the ITS assay could detect smaller amounts of DNA in a sample [
22] compared to the BCSP31. It is worth noting that BCSP31 is highly conserved in all
Brucella species and biovars, except for
Brucella ovis [
40]. It may be that this sample could be
Brucella ovis and supports the assertion that the developed ITS assay can detect any species of
Brucella.
The ITS qPCR assay produced lower C
T values than the BCSP31 assay, suggesting that the ITS assay is more sensitive than the BCSP31 assay. The difference in C
T values observed between these assays could be a result of the DNA copy number in these gene targets. The BCSP31 gene, which encodes for a 31-kDa
B. abortus antigen, is present as a single copy, whereas the 16S-23SrDNA ITS region exists as three copies [
21], and it is likely that a gene target with multiple copy numbers is more sensitive than a single copy gene.
The difference in C
T (ΔC
T) observed between the ITS and BCSP31 assays was between 7.10 in aborted foetal tissues and 3.24 in abomasal fluid. This result agrees with those of Katter et al. [
21], who found out that the C
T values were three cycles lower for ITS-PCR in tissues than that of blood. This indicated a higher bacterial load in tissues and the possible use of these assays for diagnosing the disease.
The diagnostic sensitivity and specificity of the ITS and BSCP31-qPCR was 87%/95% and 92%/89% respectively, and for culture, it was 47% and 55% respectively, which indicated that PCR is more sensitive than culture. Our ITS assay showed a greater specificity of 95% than the BCSP31 assay, with a specificity of 89%. In contrast, Katter et al. [
21] recorded the sensitivity and specificity of an ITS qPCR assay as 66.7% and 99.7% respectively, compared to culture with 77% and 100% for whole blood and paraffin-embedded tissue. Keid et al. [
22] standardised and evaluated an ITS PCR on whole blood of naturally infected dogs for the detection of
Brucella species: dogs were assigned to infected and non-infected groups based on culture and rapid slide agglutination tests, and a sensitivity and specificity of 100% was reported. The disparity in diagnostic sensitivity and specificity of PCR from various studies could be due to the use of different protocols for DNA extraction, amplification, different target genes and the choice of reference test [
22,
41].
Interestingly, the ITS assay performance, when statistically compared to BCSP31 in our study, does not reflect the theory of ‘‘an increased sensitivity corresponding to a multiple rDNA copy number’’. This could be as a result of the prior values used in determining the sensitivity of the ITS assay in the Bayesian statistical model. The priors were obtained from the prevalence, sensitivity, and specificity of the results from published literature. There is lack of information available for studies carried out on the detection of
Brucella in aborted foetal tissues using the BCSP31 gene target by qPCR. Hence, priors for the BCSP31 assay was obtained from Richtzenhain et al. [
32], which is a multiplex conventional PCR. This might have contributed to the difference in sensitivity in our result. According to Ling et al., non-identifiable latent class models are known to be greatly influenced by the subjective prior information used [
42].
In addition to the BCSP31 assay, an insertion sequence 711 (IS711)-based real-time PCR has been used to detect
Brucella spp. in blood and tissue samples in wild boars [
43]. The advantages of this assay are that IS711 is present in multiple copies in the Brucella genome and represents a stable genetic element with regard to the number and position in the genomes of various Brucella species [
8,
44]. Most species contain at least one copy of the IS element at a unique chromosomal location [
45]. The AMOS-PCR is a locus-specific multiplexing assay to differentiate Brucella species and is based on IS711 [
46].
Loop-mediated isothermal amplification (LAMP) is one of the most widely used isothermal amplification assays used for the detection of several bacterial pathogens including
Brucella sp. in clinical samples [
47,
48,
49,
50]. Since LAMP requires multiple primers, it involves rigorous optimisation and increases the chance of leftover contamination, giving false-positive results in many instances [
51]. The LAMP technique is simple because the whole process of amplification and detection is done in a single step in which the reaction components are subjected to isothermal conditions [
52]. This technique can be used on the field. It also has the advantage of working at constant temperature, results can be viewed directly without post-amplification protocols [
53].
The prevalence of brucellosis in aborted foetal tissues was 33% (19/56). This shows that prevalence has increased from initial diagnostic reports. Kolo et al. [
54] reported a prevalence of 12.5% (25/200) from screened cattle tissues by conventional ITS-PCR; however, this was conducted on abattoir samples from apparently healthy animals. In a study carried out in Egypt, a total of 47 aborted foetuses with related placenta were examined.
Brucella spp. DNA was detected in 25.5% (12/47)) of tissue samples by conventional PCR [
55].
About 41.5% of the nation’s herds were tested between 1977 and 1978. A prevalence of 6.6% was recorded. Northeastern parts of Kwa-Zulu-Natal [
56] reported a prevalence of 1.45–15.6%.
Another issue of concern is that because the current bovine brucellosis testing scheme is compulsory only for high-risk herds and voluntary for all other herds and livestock owners [
57], the status of numerous herds that were not classified as high-risk remains unknown [
57]. This leads to the assumption that the prevalence of the disease in the country is higher than previously reported [
58]. There is minimal information on brucellosis in small ruminants, pigs, and wildlife [
10].
The presence of
Brucella spp. in livestock is a major risk to public health, because livestock and animal products are the only source of infections in humans [
59].








{kind=link}
{kind=link}
{kind=link}