Nitric Oxide as a Potential Adjuvant Therapeutic for Neuroblastoma: Effects of NO on Murine N2a Cells



Abstract
1. Introduction
2. Materials and Methods
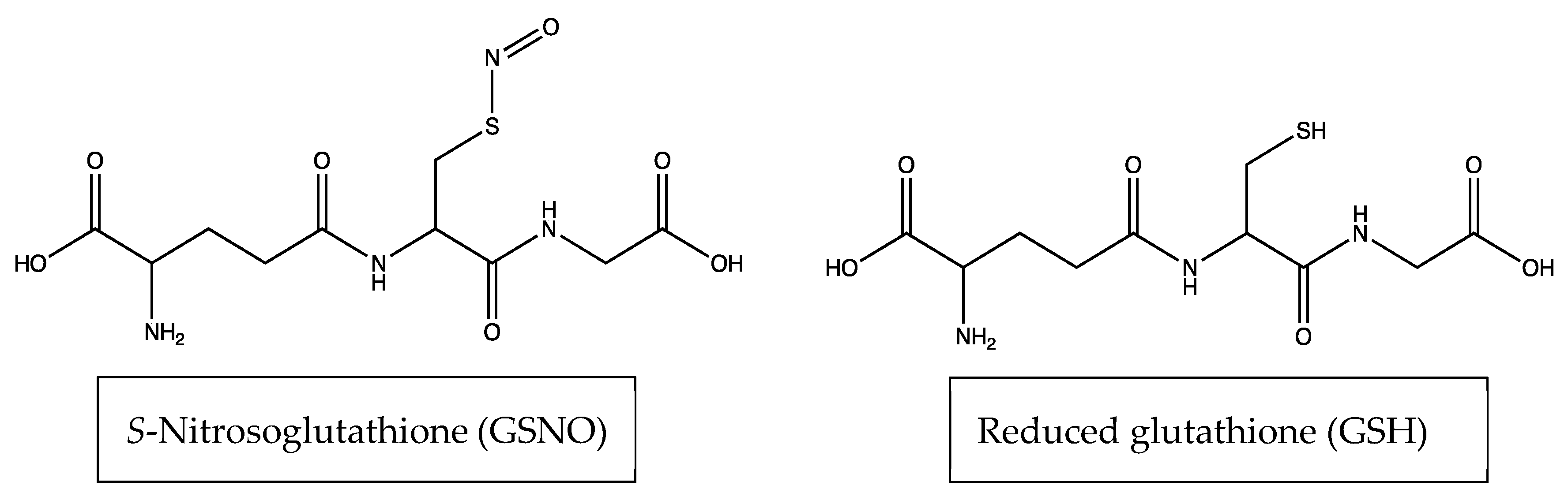
2.1. S-Nitrosoglutathione Synthesis
2.1.1. Materials
2.1.2. Synthesis of S-Nitrosoglutathione (GSNO)
2.2. Cell Culture—N2a
2.2.1. Materials
2.2.2. Method
2.3. Cell Culture—HDF
2.3.1. Materials
2.3.2. Method
2.4. In Vitro Cell Viability Assays
2.4.1. Materials
2.4.2. Method
2.4.3. Resazurin Assay
2.4.4. WST-8 Assay
2.4.5. MTT Assay
2.5. Colony Formation
Method
2.6. Nitric Oxide Analyzer Analysis of 1 mM GSNO in PBS
2.7. UV-Vis Analysis of 1 mM GSNO in Complete DMEM
2.8. Data Analysis/Statistics
3. Results
3.1. Cell Viability Assays
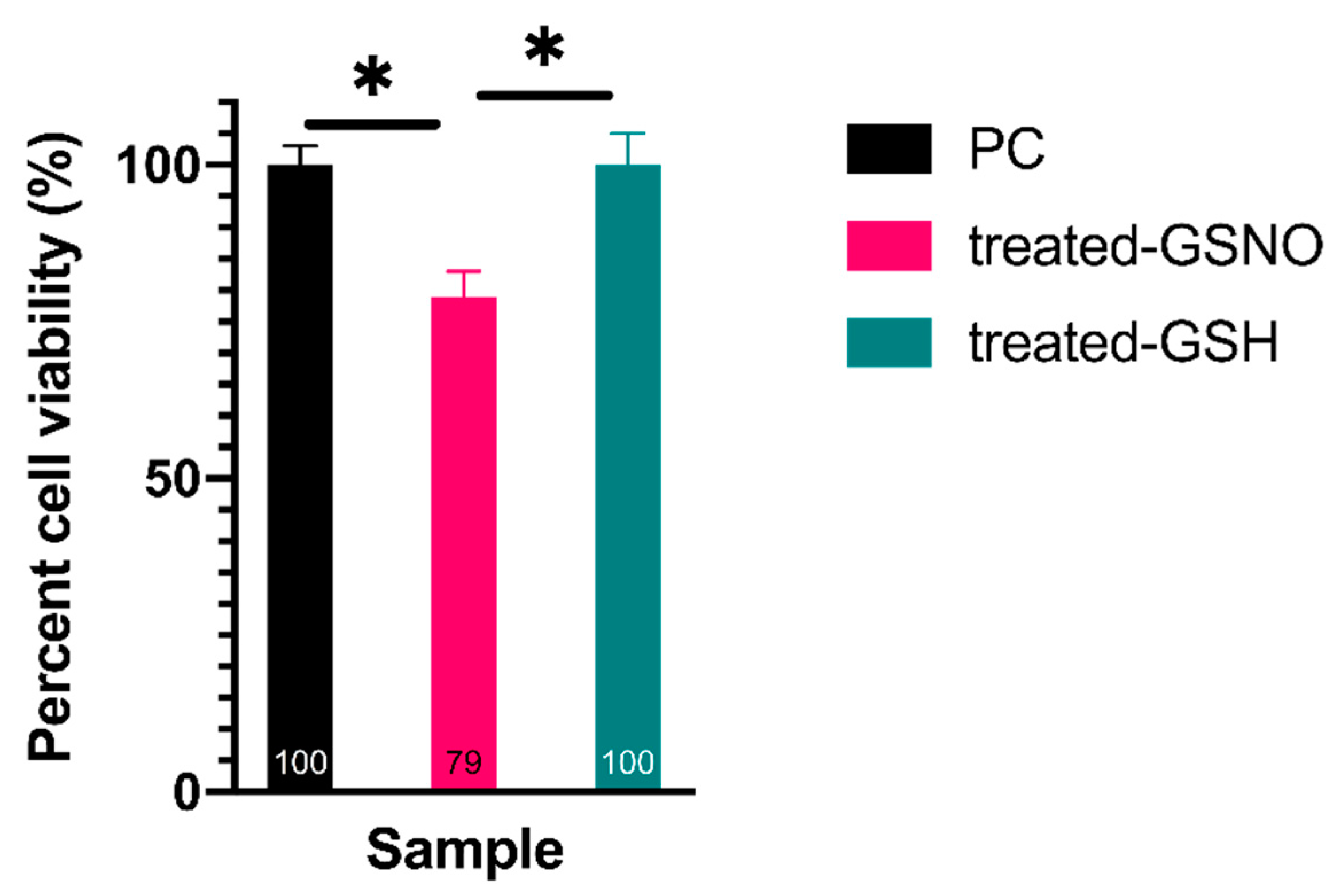
3.1.1. Resazurin Assay—Analysis of Cellular Viability
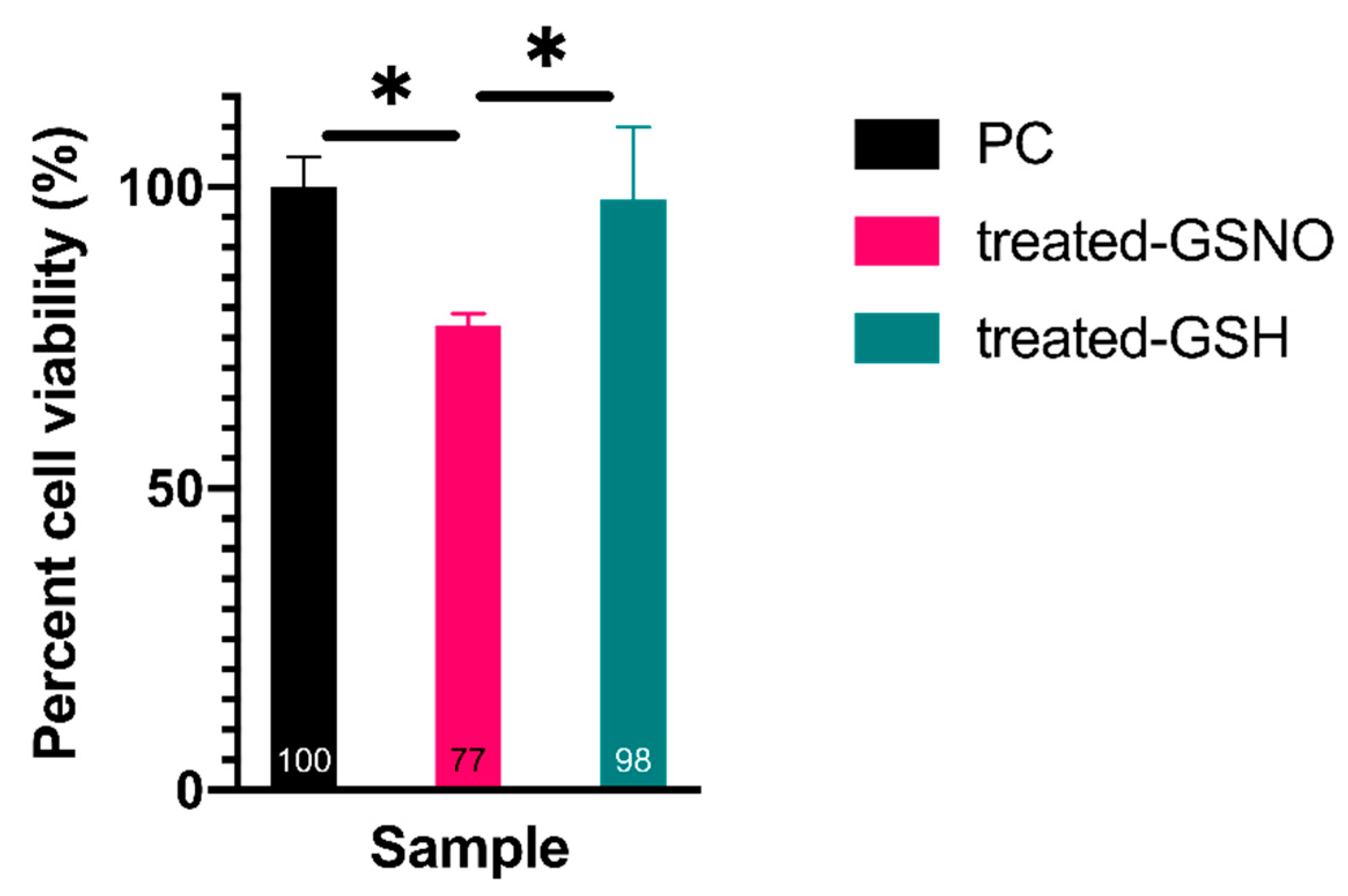
3.1.2. WST-8 Assay—Analysis of Cellular Viability
3.1.3. MTT Assay–Analysis of Cellular Viability
3.2. Colony Formation Assay
3.3. Impact of NO on HDFa Cells—Resazurin Assay
3.4. NO-Release from 1 mM GSNO
3.4.1. NO-Release from 1 mM GSNO in PBS
3.4.2. Absorbance of GSNO λmax via UV/VIS
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavalli, G. Chromatin and epigenetics in development: Blending cellular memory with cell fate plasticity. Development 2006, 133, 2089–2094. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T. The epigenetic magic of histone lysine methylation: Delivered on 6 July 2005 at the 30th FEBS Congress in Budapest, Hungary. FEBS J. 2006, 273, 3121–3135. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef]
- Esposito, M.; Sherr, G.L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 2019, 13, 1–12. [Google Scholar] [CrossRef]
- Smith, M.; Flodman, P.L. Expanded insights into mechanisms of gene expression and disease related disruptions. Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef]
- Al-Hasani, K.; Mathiyalagan, P.; El-Osta, A. Epigenetics, cardiovascular disease, and cellular reprogramming. J. Mol. Cell. Cardiol. 2019, 128, 129–133. [Google Scholar] [CrossRef]
- Ducasse, M.; Brown, M.A. Epigenetic aberrations and cancer. Mol. Cancer 2005, 5, 1–10. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2012, 23, 1–7. [Google Scholar] [CrossRef]
- Hayat, M. Neuroblastoma; Hayat, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–11. [Google Scholar] [CrossRef]
- Shohet, J.; Foster, J. Neuroblastoma. Br. Med. J. 2017, 357, j1863. [Google Scholar] [CrossRef]
- Brodeur, B.G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.T.; Castel, V.; Castleberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Matthay, K.K.; Atkinson, J.B.; Seeger, R.C.; Shimada, H.; Haase, G.M.; Stram, D.O.; Gerbing, R.B.; Lukens, J.N. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: A children’s cancer group study. J. Clin. Oncol. 2000, 18, 18–26. [Google Scholar] [PubMed]
- Nuchtem, J.G.; London, W.B.; Barnewolt, C.E.; Naranjo, A.; McGrady, P.W. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants, a children’s oncology group study. Ann. Surg. 2017, 256, 573–580. [Google Scholar] [CrossRef]
- Pe, D.; Le, C.; Oscarlambret, C.; Dieu, H. Localised and unresectable neuroblastoma in infants: Excellent outcome with low-dose primary chemotherapy. Br. J. Cancer 2003, 89, 1605–1609. [Google Scholar] [CrossRef][Green Version]
- Nickerson, B.H.J.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; Shimada, H.; Perez, C.; Atkinson, J.B.; Selch, M.; Gerbing, R.B.; Stram, D.O.; et al. Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: A children’ s cancer group study results. J. Clin. Oncol. 2000, 18, 477–486. [Google Scholar] [CrossRef]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkins, E.S.; Haas-Kogan, D.; Gerbing, R.B.; London, W.B.; Villablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Ash, S.; Laureys, G.; Brock, P.R.; Michon, J.M.; Owens, C.; et al. Investigation of the role of dinutuximab beta-based immunotherapy in the siopen high-risk neuroblastoma 1 trial (HR-NBL1). Cancers 2020, 12, 309. [Google Scholar] [CrossRef]
- Oh-hashi, K.; Maruyama, W.; Yi, H.; Takahashi, T.; Naoi, M.; Isobe, K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 1999, 1999, 504–509. [Google Scholar]
- Kim, B.-C.; Kim, Y.-S.; Lee, J.-W.; Seo, J.-H.; Ji, E.-S.; Lee, H.; Park, Y.-I.; Kim, C.-J. Protective effect of coriolus versicolor cultivated in citrus extract against nitric oxide-induced apoptosis in human neuroblastoma SK-N-MC cells. Exp. Neurobiol. 2011, 20, 100. [Google Scholar] [CrossRef]
- Korde Choudhari, S.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Stevens, E.V.; Carpenter, A.W.; Shin, J.H.; Liu, J.; Der, C.J.; Schoenfisch, M.H. Nitric oxide-releasing silica nanoparticle inhibition of ovarian cancer cell growth. Mol. Pharm. 2010, 7, 775–785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suchyta, D.J.; Schoenfisch, M.H. Anticancer potency of nitric oxide-releasing liposomes. RSC Adv. 2017, 7, 53236–53246. [Google Scholar] [CrossRef] [PubMed]
- Kogias, E.; Osterberg, N.; Baumer, B.; Psarras, N.; Koentges, C.; Papazoglou, A.; Saavedra, J.E.; Keefer, L.K.; Weyerbrock, A. Growth-inhibitory and chemosensitizing effects of the glutathione-S- transferase-π-activated nitric oxide donor PABA/NO in malignant gliomas. Int. J. Cancer 2012, 130, 1184–1194. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Saavedra, J.E.; Buzard, G.S.; Citro, M.L.; Keefer, L.K.; Ji, X.; Tew, K.D. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol. 2004, 65, 1070–1079. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, J.; Andreana, P.R.; Cantauria, G.; Tarasia, S.; Sharp, L.; Braunschweiger, P.G.; Wang, P.G. Targeting nitric oxide to cancer cells: Cytotoxicity studies of glyco-S- nitrosothiols. Bioorg. Med. Chem. Lett. 1999, 9, 2255–2258. [Google Scholar] [CrossRef]
- Dong, R.; Wang, X.; Wang, H.; Liu, Z.; Liu, J.; Saavedra, J.E. Effects of JS-K, a novel anti-cancer nitric oxide prodrug, on gene expression in human hepatoma Hep3B cells. Biomed. Pharmacother. 2017, 88, 367–373. [Google Scholar] [CrossRef]
- Reynolds, M.M.; Witzeling, S.D.; Damodaran, V.B.; Medeiros, T.N.; Knodle, R.D.; Edwards, M.A.; Lookian, P.P.; Brown, M.A. Applications for nitric oxide in halting proliferation of tumor cells. Biochem. Biophys. Res. Commun. 2013, 431, 647–651. [Google Scholar] [CrossRef]
- Nortcliffe, A.; Ekstrom, A.G.; Black, J.R.; Ross, J.A.; Habib, F.K.; Botting, N.P.; O’Hagan, D. Synthesis and biological evaluation of nitric oxide-donating analogues of sulindac for prostate cancer treatment. Bioorg. Med. Chem. 2014, 22, 756–761. [Google Scholar] [CrossRef]
- Lee, S.Y.; Rim, Y.; McPherson, D.D.; Huang, S.L.; Kim, H. A novel liposomal nanomedicine for nitric oxide delivery and breast cancer treatment. Biomed. Mater. Eng. 2014, 24, 61–67. [Google Scholar] [CrossRef]
- Fu, J.; Han, J.; Meng, T.; Hu, J.; Yin, J. Novel α-ketoamide based diazeniumdiolates as hydrogen peroxide responsive nitric oxide donors with anti-lung cancer activity. Chem. Commun. 2019, 55, 12904–12907. [Google Scholar] [CrossRef] [PubMed]
- Suchyta, D.J.; Schoenfisch, M.H. Encapsulation of N-diazeniumdiolates within liposomes for enhanced nitric oxide donor stability and delivery. Mol. Pharm. 2015, 12, 3569–3574. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M. Chemistry of nitric oxide: Biologically relevant aspects. Adv. Pharmacol. 1995, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide Biol. Chem. 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Yang, D.I.; Yin, J.H.; Ju, T.C.; Chen, L.S.; Hsu, C.Y. Nitric oxide and BCNU chemoresistance in C6 glioma cells: Role of S-Nitrosoglutathione. Free Radic. Biol. Med. 2004, 36, 1317–1328. [Google Scholar] [CrossRef]
- Müerköster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1β. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef]
- Safdar, S.; Payne, C.A.; Tu, N.H.; Taite, L.J. Targeted nitric oxide delivery preferentially induces glioma cell chemosensitivity via altered p53 and O6-Methylguanine-DNA Methyltransferase activity. Biotechnol. Bioeng. 2013, 110, 1211–1220. [Google Scholar] [CrossRef]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Li, L.; Zhu, L.; Hao, B.; Gao, W.; Wang, Q.; Li, K.; Wang, M.; Huang, M.; Liu, Z.; Yang, Q.; et al. iNOS-derived nitric oxide promotes glycolysis by inducing pyruvate kinase M2 nuclear translocation in ovarian cancer. Oncotarget 2017, 8, 33047–33063. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Absorbance at 336 nm (GSNO λmax) | Concentration of GSNO (mM) | Amount of Available NO (μmol) |
|---|---|---|---|
| 0 h | 0.895 | 0.971 | 0.433 |
| 6 h | 0.598 | 0.649 | 0.290 |
| 12 h | 0.304 | 0.330 | 0.147 |
| 18 h | 0.130 | 0.141 | 0.0630 |
| 24 h | 0.135 | 0.146 | 0.0650 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, J.L.; Reynolds, M.M.; Brown, M.A. Nitric Oxide as a Potential Adjuvant Therapeutic for Neuroblastoma: Effects of NO on Murine N2a Cells. Vet. Sci. 2020, 7, 51. https://doi.org/10.3390/vetsci7020051
Gordon JL, Reynolds MM, Brown MA. Nitric Oxide as a Potential Adjuvant Therapeutic for Neuroblastoma: Effects of NO on Murine N2a Cells. Veterinary Sciences. 2020; 7(2):51. https://doi.org/10.3390/vetsci7020051
Chicago/Turabian StyleGordon, Jenna L., Melissa M. Reynolds, and Mark A. Brown. 2020. "Nitric Oxide as a Potential Adjuvant Therapeutic for Neuroblastoma: Effects of NO on Murine N2a Cells" Veterinary Sciences 7, no. 2: 51. https://doi.org/10.3390/vetsci7020051
APA StyleGordon, J. L., Reynolds, M. M., & Brown, M. A. (2020). Nitric Oxide as a Potential Adjuvant Therapeutic for Neuroblastoma: Effects of NO on Murine N2a Cells. Veterinary Sciences, 7(2), 51. https://doi.org/10.3390/vetsci7020051