
Feline Immunodeficiency Virus Neuropathogenesis: A Model for HIV-Induced CNS Inflammation and Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. FIV Infection of the Nervous System
2.1. General Background
2.2. FIV Trafficking across the Blood-Brain Barrier
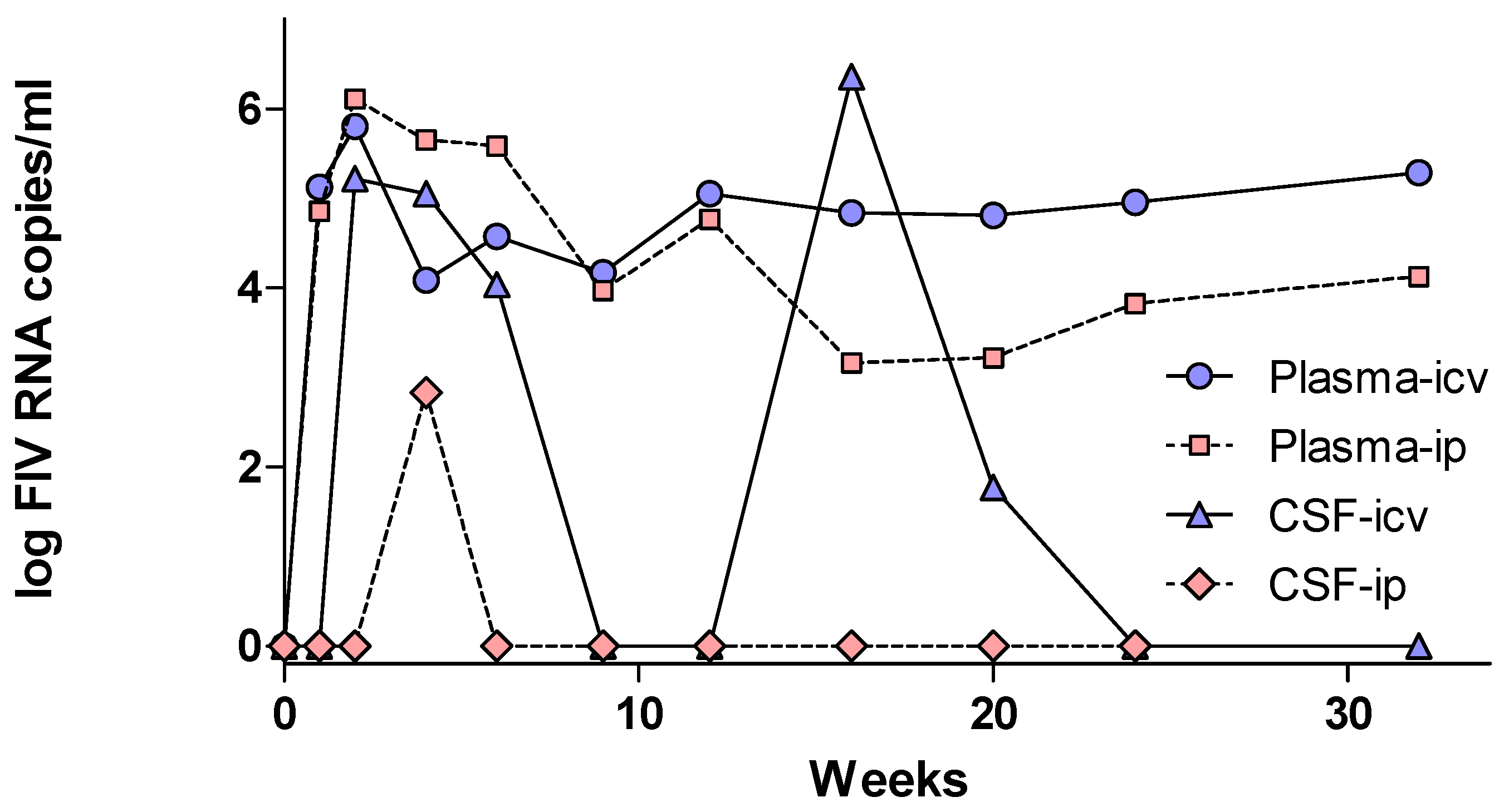
2.3. FIV Trafficking across the Blood–CSF Barrier
3. FIV and Neuropathogenesis
3.1. Neurological Signs
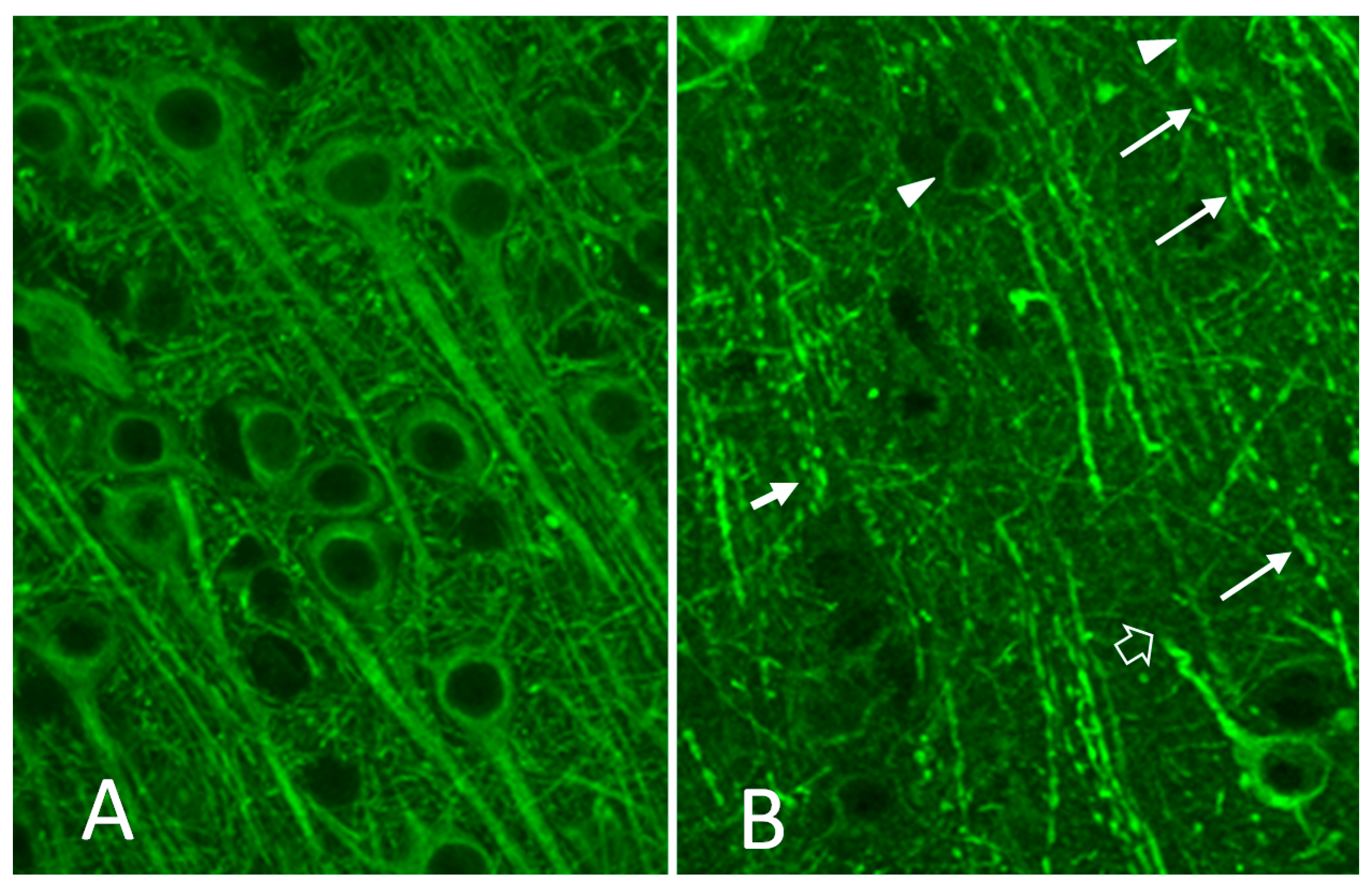
3.2. FIV Neuropathology
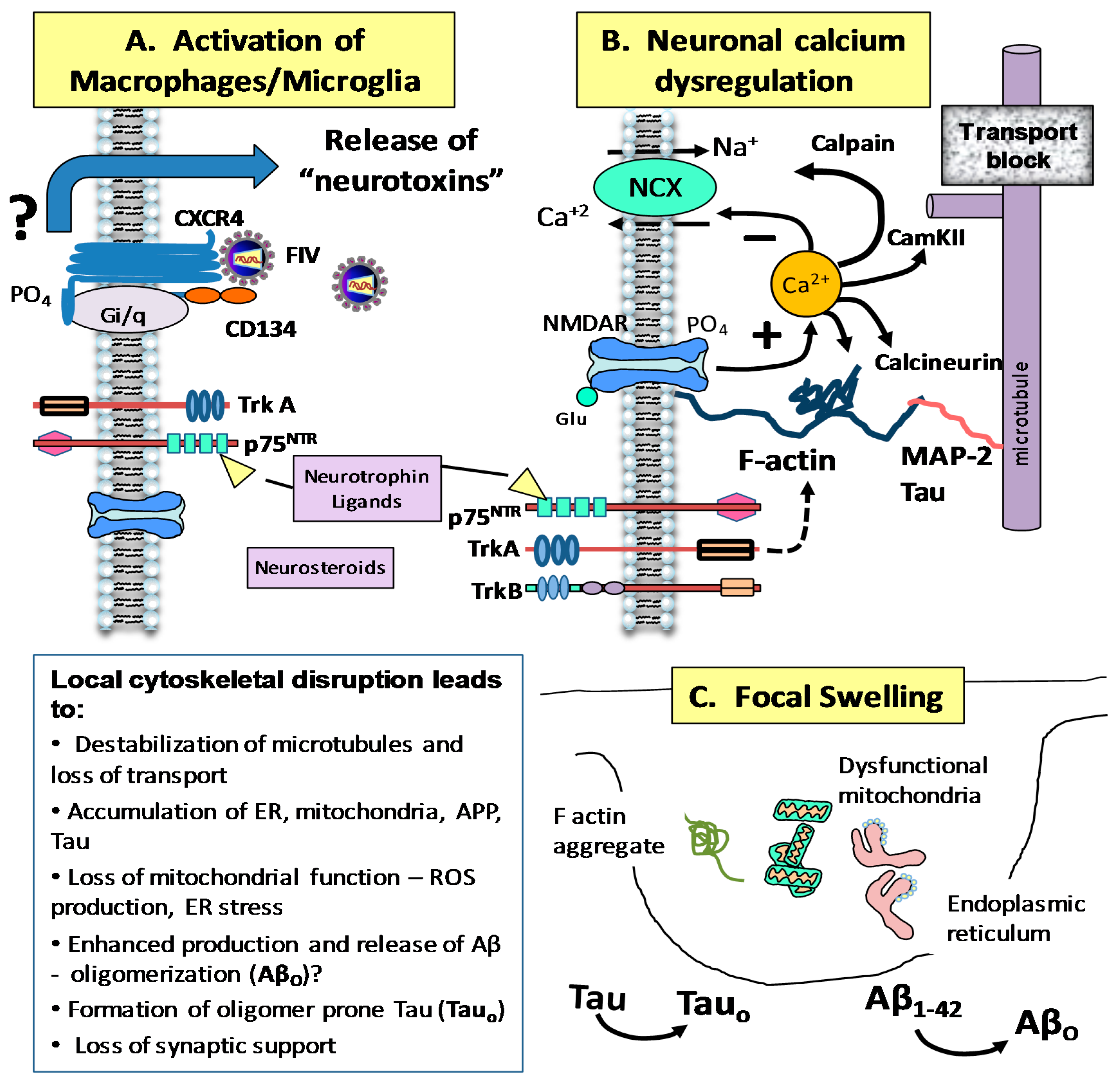
4. Mechanisms of FIV Neurotoxicity
4.1. Studies Using Cultured Fetal Feline Cells
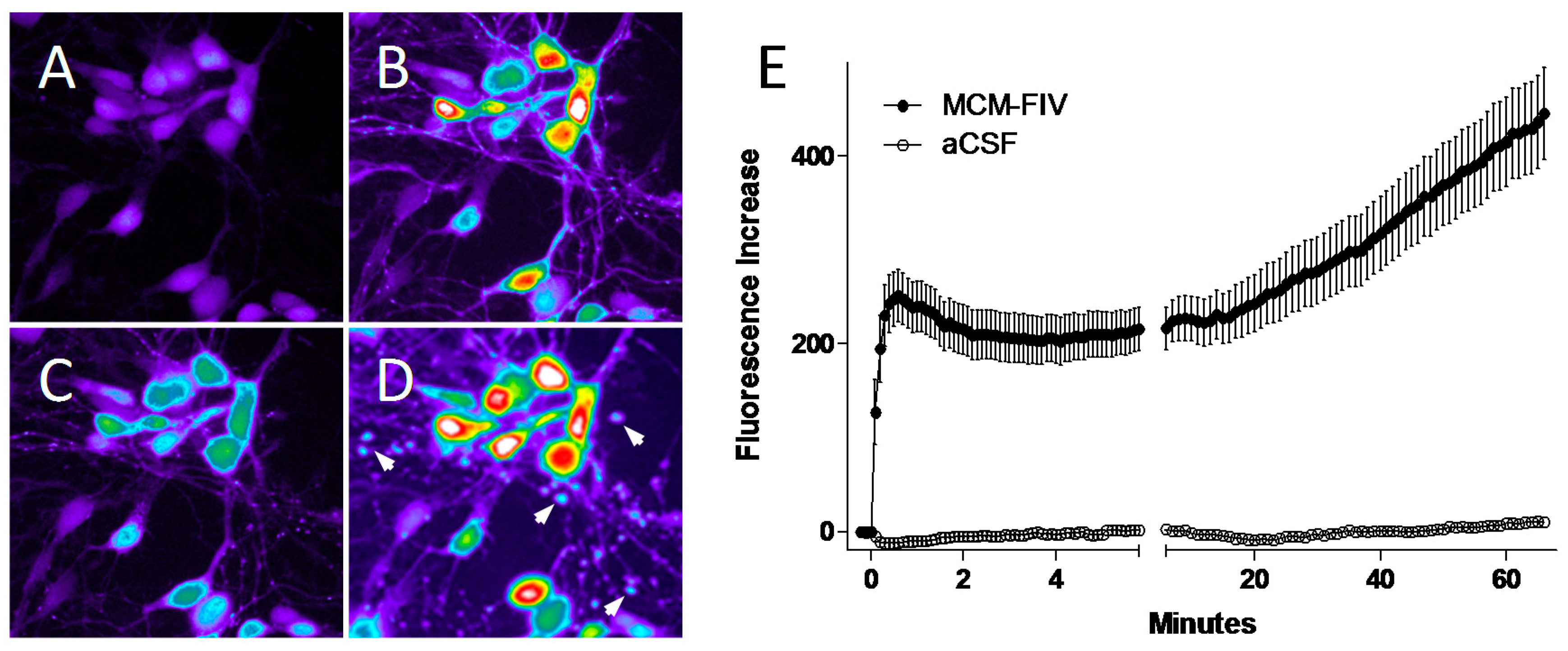
4.2. FIV and Intracellular Calcium Homeostasis
5. Development of Neuroprotective Treatments
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brinkmann, R.; Schwinn, A.; Narayan, O.; Zink, C.; Kreth, H.W.; Roggendorf, W.; Dorries, R.; Schwender, S.; Imrich, H.; Ter, M.V. Human immunodeficiency virus infection in microglia: Correlation between cells infected in the brain and cells cultured from infectious brain tissue. Ann. Neurol. 1992, 31, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.W.; Mathiason, C.K.; Hoover, E.A. In vivo monocyte tropism of pathogenic feline immunodeficiency viruses. J. Virol. 1999, 73, 6852–6861. [Google Scholar] [PubMed]
- Fischer-Smith, T.; Croul, S.; Sverstiuk, A.E.; Capini, C.; L’Heureux, D.; Regulier, E.G.; Richardson, M.W.; Amini, S.; Morgello, S.; Khalili, K.; et al. Cns invasion by cd14+/cd16+ peripheral blood-derived monocytes in hiv dementia: Perivascular accumulation and reservoir of hiv infection. J. NeuroVirol. 2001, 7, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of aids. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.R.; Bristol, G.; Zack, J.A.; Ritola, K.; Swanstrom, R.; Birch, C.J.; Bell, J.E.; Bannert, N.; Crawford, K.; Wang, H.; et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J. Virol. 2001, 75, 10073–10089. [Google Scholar] [CrossRef] [PubMed]
- Hein, A.; Martin, J.P.; Koehren, F.; Bingen, A.; Dorries, R. In vivo infection of ramified microglia from adult cat central nervous system by feline immunodeficiency virus. Virology 2000, 268, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; Dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of aids virus in macrophages in brain tissue from aids patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.H.; Sasseville, V.G.; Smith, M.O.; Vogel, P.; Pauley, D.R.; Heyes, M.P.; Lackner, A.A. Neuroinvasion by simian immunodeficiency virus coincides with increased numbers of perivascular macrophages/microglia and intrathecal immune activation. J. NeuroVirol. 1996, 2, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J. NeuroVirol. 2004, 10, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Corey, S.; Westmoreland, S.V.; Pauley, D.; Knight, H.; deBakker, C.; Alvarez, X.; Lackner, A.A. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: Implications for the neuropathogenesis of aids. J. Exp. Med. 2001, 193, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Zenger, E.; Tiffany-Castiglioni, E.; Collisson, E.W. Cellular mechanisms of feline immunodeficiency virus (FIV)-induced neuropathogenesis. Front. Biosci. 1997, 2, d527–d537. [Google Scholar] [CrossRef] [PubMed]
- Anthony, I.C.; Ramage, S.N.; Carnie, F.W.; Simmonds, P.; Bell, J.E. Influence of haart on HIV-related CNS disease and neuroinflammation. J. Neuropathol. Exp. Neurol. 2005, 64, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Neuenburg, J.K.; Brodt, H.R.; Herndier, B.G.; Bickel, M.; Bacchetti, P.; Price, R.W.; Grant, R.M.; Schlote, W. Hiv-related neuropathology, 1985 to 1999: Rising prevalence of hiv encephalopathy in the era of highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2002, 31, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, N.; McDermott, M.P.; Marder, K.; Schifitto, G.; Selnes, O.A.; McArthur, J.C.; Stern, Y.; Albert, S.; Palumbo, D.; Kieburtz, K.; et al. Hiv-associated cognitive impairment before and after the advent of combination therapy. J. NeuroVirol. 2002, 8, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. NeuroVirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Cysique, L.A.; Brew, B.J.; Halman, M.; Catalan, J.; Sacktor, N.; Price, R.W.; Brown, S.; Atkinson, J.H.; Clifford, D.B.; Simpson, D.; et al. Undetectable cerebrospinal fluid hiv rna and beta-2 microglobulin do not indicate inactive aids dementia complex in highly active antiretroviral therapy-treated patients. J. Acquir. Immune Defic. Syndr. 2005, 39, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect Biol. 2015, 7, a020537. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2016, 126, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Ho, E.W.; Brown, M.L.; Yamamoto, J.K. Isolation of a t-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 1987, 235, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B. Feline immunodeficiency virus neuropathogenesis: From cats to calcium. J. Neuroimmune Pharmacol. 2007, 2, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Moench, T.R. Cell-associated transmission of HIV type 1 and other lentiviruses in small-animal models. J. Infect. Dis. 2014, 210, S654–S659. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Hosie, M.J. The virus-receptor interaction in the replication of feline immunodeficiency virus (FIV). Curr. Opin. Virol. 2013, 3, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Kraase, M.; Logan, N.; McMonagle, E.; Varela, M.; Hosie, M.J. Selective expansion of viral variants following experimental transmission of a reconstituted feline immunodeficiency virus quasispecies. PLoS One 2013, 8, e54871. [Google Scholar]
- Beczkowski, P.M.; Hughes, J.; Biek, R.; Litster, A.; Willett, B.J.; Hosie, M.J. Rapid evolution of the ENV gene leader sequence in cats naturally infected with feline immunodeficiency virus. J. Gen. Virol. 2015, 96, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Beczkowski, P.M.; Techakriengkrai, N.; Logan, N.; McMonagle, E.; Litster, A.; Willett, B.J.; Hosie, M.J. Emergence of cd134 cysteine-rich domain 2 (crd2)-independent strains of feline immunodeficiency virus (FIV) is associated with disease progression in naturally infected cats. Retrovirology 2014, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, R.; Izumi, T.; Yamada, E.; Nakano, Y.; Misawa, N.; Ren, F.; Carpenter, M.A.; Ikeda, T.; Munk, C.; Harris, R.S.; et al. A naturally occurring domestic cat apobec3 variant confers resistance to feline immunodeficiency virus infection. J. Virol. 2015, 90, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Beczkowski, P.M.; Logan, N.; McMonagle, E.; Litster, A.; Willett, B.J.; Hosie, M.J. An investigation of the breadth of neutralizing antibody response in cats naturally infected with feline immunodeficiency virus. J. Gen. Virol. 2015, 96, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Westman, M.E.; Malik, R.; Hall, E.; Harris, M.; Norris, J.M. The protective rate of the feline immunodeficiency virus vaccine: An Australian field study. Vaccine 2016, 34, 4752–4758. [Google Scholar] [CrossRef] [PubMed]
- Beczkowski, P.M.; Harris, M.; Techakriengkrai, N.; Beatty, J.A.; Willett, B.J.; Hosie, M.J. Neutralising antibody response in domestic cats immunised with a commercial feline immunodeficiency virus (FIV) vaccine. Vaccine 2015, 33, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.K.; Pu, R.; Martin, M.M.; Noon-Song, E.N.; Zwijnenberg, R.; Yamamoto, J.K. Feline immunodeficiency virus (FIV) vaccine efficacy and FIV neutralizing antibodies. Vaccine 2014, 32, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Guevara, R.B.; Marcano, A.C.; Saenz, D.T.; Fadel, H.J.; Rogstad, D.K.; Poeschla, E.M. Feline immunodeficiency virus envelope glycoproteins antagonize tetherin through a distinctive mechanism that requires virion incorporation. J. Virol. 2014, 88, 3255–3272. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Cox, C.; Baptiste, J.; Summers, H.; Button, R.; Bahlow, K.; Spurrier, V.; Kyser, J.; Luttge, B.G.; Kuo, L.; et al. Nmr structure of the myristylated feline immunodeficiency virus matrix protein. Viruses 2015, 7, 2210–2229. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, S.J.; Sparger, E.E.; Luciw, P.A.; Murphy, B.G. Pharmacologic reactivation of latent feline immunodeficiency virus ex vivo in peripheral cd4+ t-lymphocytes. Virus Res. 2012, 170, 174–179. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, S.J.; Sparger, E.E.; Murphy, B.G. Feline immunodeficiency virus latency. Retrovirology 2013, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Vapniarsky, N.; Hillman, C.; Castillo, D.; McDonnel, S.; Moore, P.; Luciw, P.A.; Sparger, E.E. FIV establishes a latent infection in feline peripheral blood cd4+ t lymphocytes in vivo during the asymptomatic phase of infection. Retrovirology 2012, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.S.; VanLare, K.A.; Roycroft, L.M.; King, S. Effect of high-dose ciclosporin on the immune response to primary and booster vaccination in immunocompetent cats. J. Feline Med. Surg. 2015, 17, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Gil, S.; Leal, R.O.; McGahie, D.; Sepulveda, N.; Duarte, A.; Niza, M.M.; Tavares, L. Oral recombinant feline interferon-omega as an alternative immune modulation therapy in FIV positive cats: Clinical and laboratory evaluation. Res. Vet. Sci. 2014, 96, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S.; Duarte, A.; McGahie, D.; Sepulveda, N.; Niza, M.M.; Tavares, L. Evaluation of viremia, proviral load and cytokine profile in naturally feline immunodeficiency virus infected cats treated with two different protocols of recombinant feline interferon omega. Res. Vet. Sci. 2015, 99, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S.; Sepulveda, N.; McGahie, D.; Duarte, A.; Niza, M.M.; Tavares, L. Monitoring acute phase proteins in retrovirus infected cats undergoing feline interferon-omega therapy. J. Small Anim. Pract. 2014, 55, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Tompkins, M.; Miller, M.; Fogle, J. Lentivirus-activated t regulatory cells suppress t helper cell interleukin-2 production by inhibiting nuclear factor of activated t cells 2 binding to the interleukin-2 promoter. AIDS Res. Hum. Retrovir. 2014, 30, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Akaronu, N.; Thompson, E.M.; Hood, S.F.; Fogle, J.E. Modulating DNA methylation in activated cd8+ t cells inhibits regulatory t cell-induced binding of foxp3 to the cd8+ t cell il-2 promoter. J. Immunol. 2015, 194, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Fogle, J.E.; Tompkins, M.B. Infection with feline immunodeficiency virus directly activates CD4+ CD25+ t regulatory cells. J. Virol. 2013, 87, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Petty, C.S.; Tompkins, M.B.; Fogle, J.E. CD4+CD25+ t regulatory cells activated during feline immunodeficiency virus infection convert t helper cells into functional suppressors through a membrane-bound tgfbeta / garp-mediated mechanism. Virol. J. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reckling, S.; Dean, G.A. Phenotypic and functional analysis of cd1a+ dendritic cells from cats chronically infected with feline immunodeficiency virus. Comp. Immunol. Microbiol. Infect. Dis. 2015, 42, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Weese, J.S.; Nichols, J.; Jalali, M.; Litster, A. The rectal microbiota of cats infected with feline immunodeficiency virus infection and uninfected controls. Vet. Microbiol. 2015, 180, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Branton, W.G.; Ellestad, K.K.; Maingat, F.; Wheatley, B.M.; Rud, E.; Warren, R.L.; Holt, R.A.; Surette, M.G.; Power, C. Brain microbial populations in hiv/aids: Alpha-proteobacteria predominate independent of host immune status. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Asquith, C.R.; Meli, M.L.; Konstantinova, L.S.; Laitinen, T.; Perakyla, M.; Poso, A.; Rakitin, O.A.; Allenspach, K.; Hofmann-Lehmann, R.; Hilton, S.T. Evaluation of the antiviral efficacy of bis[1,2]dithiolo[1,4]thiazines and bis[1,2]dithiolopyrrole derivatives against the nucelocapsid protein of the feline immunodeficiency virus (FIV) as a model for HIV infection. Bioorg. Med. Chem. Lett. 2014, 24, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.; Meli, M.L.; Konstantinova, L.S.; Laitinen, T.; Poso, A.; Rakitin, O.A.; Hofmann-Lehmann, R.; Allenspach, K.; Hilton, S.T. Novel fused tetrathiocines as antivirals that target the nucleocapsid zinc finger containing protein of the feline immunodeficiency virus (FIV) as a model of hiv infection. Bioorg. Med. Chem. Lett. 2015, 25, 1352–1355. [Google Scholar] [CrossRef]
- Taffin, E.; Paepe, D.; Goris, N.; Auwerx, J.; Debille, M.; Neyts, J.; Van de Maele, I.; Daminet, S. Antiviral treatment of feline immunodeficiency virus-infected cats with (r)-9-(2-phosphonylmethoxypropyl)-2,6-diaminopurine. J. Feline Med. Surg. 2015, 17, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.L.; Gruen, M.E.; Meeker, R.B.; Milgram, B.; DiRivera, C.; Thomson, A.; Clary, G.; Hudson, L. The use of a T-maze to measure cognitive-motor function in cats (Felis catus). J. Vet. Behav. 2013, 8, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Maingat, F.; Vivithanaporn, P.; Zhu, Y.; Taylor, A.; Baker, G.; Pearson, K.; Power, C. Neurobehavioral performance in feline immunodeficiency virus infection: Integrated analysis of viral burden, neuroinflammation, and neuronal injury in cortex. J. Neurosci. 2009, 29, 8429–8437. [Google Scholar] [CrossRef] [PubMed]
- Maingat, F.G.; Polyak, M.J.; Paul, A.M.; Vivithanaporn, P.; Noorbakhsh, F.; Ahboucha, S.; Baker, G.B.; Pearson, K.; Power, C. Neurosteroid-mediated regulation of brain innate immunity in HIV/AIDs: Dhea-s suppresses neurovirulence. FASEB J. 2013, 27, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Steigerwald, E.S.; Sarter, M.; March, P.; Podell, M. Effects of feline immunodeficiency virus on cognition and behavioral function in cats. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1999, 20, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Bienzle, D. Fiv in cats—A useful model of HIV in people? Vet. Immunol. Immunopathol. 2014, 159, 171–179. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A.; Chatterji, U.; Sun, P.; Elder, J.H. Feline immunodeficiency virus targets activated cd4+ t cells by using cd134 as a binding receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 13044–13049. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Miyazawa, T.; Ikeda, Y.; McMonagle, E.L.; Haining, H.; Akashi, H.; Takeuchi, Y.; Hosie, M.J.; Willett, B.J. Use of cd134 as a primary receptor by the feline immunodeficiency virus. Science 2004, 303, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Egberink, H.F.; De Clercq, E.; van Vliet, A.L.; Balzarini, J.; Bridger, G.J.; Henson, G.; Horzinek, M.C.; Schols, D. Bicyclams, selective antagonists of the human chemokine receptor cxcr4, potently inhibit feline immunodeficiency virus replication. J. Virol. 1999, 73, 6346–6352. [Google Scholar] [PubMed]
- Hosie, M.J.; Broere, N.; Hesselgesser, J.; Turner, J.D.; Hoxie, J.A.; Neil, J.C.; Willett, B.J. Modulation of feline immunodeficiency virus infection by stromal cell-derived factor. J. Virol. 1998, 72, 2097–2104. [Google Scholar] [PubMed]
- Willett, B.J.; Adema, K.; Heveker, N.; Brelot, A.; Picard, L.; Alizon, M.; Turner, J.D.; Hoxie, J.A.; Peiper, S.; Neil, J.C.; et al. The second extracellular loop of cxcr4 determines its function as a receptor for feline immunodeficiency virus. J. Virol. 1998, 72, 6475–6481. [Google Scholar] [PubMed]
- Willett, B.J.; Picard, L.; Hosie, M.J.; Turner, J.D.; Adema, K.; Clapham, P.R. Shared usage of the chemokine receptor cxcr4 by the feline and human immunodeficiency viruses. J. Virol. 1997, 71, 6407–6415. [Google Scholar] [PubMed]
- Richardson, J.; Pancino, G.; Merat, R.; Leste-Lasserre, T.; Moraillon, A.; Schneider-Mergener, J.; Alizon, M.; Sonigo, P.; Heveker, N. Shared usage of the chemokine receptor cxcr4 by primary and laboratory-adapted strains of feline immunodeficiency virus. J. Virol. 1999, 73, 3661–3671. [Google Scholar] [PubMed]
- Beebe, A.M.; Dua, N.; Faith, T.G.; Moore, P.F.; Pedersen, N.C.; Dandekar, S. Primary stage of feline immunodeficiency virus infection: Viral dissemination and cellular targets. J. Virol. 1994, 68, 3080–3091. [Google Scholar] [PubMed]
- Brunner, D.; Pedersen, N.C. Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J. Virol. 1989, 63, 5483–5488. [Google Scholar] [PubMed]
- Dow, S.W.; Dreitz, M.J.; Hoover, E.A. Feline immunodeficiency virus neurotropism: Evidence that astrocytes and microglia are the primary target cells. Vet. Immunol. Immunopathol. 1992, 35, 23–35. [Google Scholar] [CrossRef]
- Bucci, J.G.; English, R.V.; Jordan, H.L.; Childers, T.A.; Tompkins, M.B.; Tompkins, W.A. Mucosally transmitted feline immunodeficiency virus induces a CD8+ antiviral response that correlates with reduction of cell-associated virus. J. Infect. Dis. 1998, 177, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, D.H.; Dow, J.L.; Childers, T.A.; Alvelo, J.I.; Tompkins, M.B.; Tompkins, W.A. Progressive expansion of an L-selectin-negative CD8 cell with anti-feline immunodeficiency virus (FIV) suppressor function in the circulation of FIV-infected cats. J. Infect. Dis. 1999, 180, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Hohdatsu, T.; Miyagawa, N.; Ohkubo, M.; Kida, K.; Koyama, H. Studies on feline cd8+ t cell non-cytolytic anti-feline immunodeficiency virus (FIV) activity. Arch. Virol. 2000, 145, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Hohdatsu, T.; Sasagawa, T.; Yamazaki, A.; Motokawa, K.; Kusuhara, H.; Kaneshima, T.; Koyama, H. Cd8+ t cells from feline immunodeficiency virus (FIV) infected cats suppress exogenous fiv replication of their peripheral blood mononuclear cells in vitro. Arch. Virol. 2002, 147, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.W.; Poss, M.L.; Hoover, E.A. Feline immunodeficiency virus: A neurotropic lentivirus. J. Acquir. Immune Defic. Syndr. 1990, 3, 658–668. [Google Scholar] [PubMed]
- Power, C.; Moench, T.; Peeling, J.; Kong, P.A.; Langelier, T. Feline immunodeficiency virus causes increased glutamate levels and neuronal loss in brain. Neuroscience 1997, 77, 1175–1185. [Google Scholar] [CrossRef]
- Macchi, S.; Maggi, F.; Di Iorio, C.; Poli, A.; Bendinelli, M.; Pistello, M. Detection of feline immunodeficiency proviral sequences in lymphoid tissues and the central nervous system by in situ gene amplification. J. Virol. Methods 1998, 73, 109–119. [Google Scholar] [CrossRef]
- Johnston, J.; Power, C. Productive infection of human peripheral blood mononuclear cells by feline immunodeficiency virus: Implications for vector development. J. Virol. 1999, 73, 2491–2498. [Google Scholar] [PubMed]
- Billaud, J.N.; Selway, D.; Yu, N.; Phillips, T.R. Replication rate of feline immunodeficiency virus in astrocytes is envelope dependent: Implications for glutamate uptake. Virology 2000, 266, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Maeda, K.; Tohya, Y.; Furuya, T.; Miyazawa, T.; Horimoto, T.; Norimine, J.; Kai, C.; Mikami, T. Replicative difference in early-passage feline brain cells among feline immunodeficiency virus isolates. Arch. Virol. 1992, 125, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Billaud, J.N.; Phillips, T.R. Effects of feline immunodeficiency virus on astrocyte glutamate uptake: Implications for lentivirus-induced central nervous system diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 2624–2629. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.; Childers, T.; Tompkins, M.; Tompkins, W.; Meeker, R. Infection of the choroid plexus by feline immunodeficiency virus. J. NeuroVirol. 2002, 8, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Steffan, A.M.; Lafon, M.E.; Gendrault, J.L.; Koehren, K.; DeMonte, M.; Royer, C.; Kirn, A.; Gut, J.P. Feline immunodeficiency virus can productively infect cultured endothelial cells from cat brain microvessels. J. Gen. Virol. 1994, 75, 3647–3653. [Google Scholar] [CrossRef] [PubMed]
- Hein, A.; Schuh, H.; Thiel, S.; Martin, J.P.; Dorries, R. Ramified feline microglia selects for distinct variants of feline immunodeficiency virus during early central nervous system infection. J. NeuroVirol. 2003, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Hudson, L.C.; Tompkins, M.B.; Vahlenkamp, T.W.; Meeker, R.B. Compartmentalization and evolution of feline immunodeficiency virus between the central nervous system and periphery following intracerebroventricular or systemic inoculation. J. NeuroVirol. 2006, 12, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Power, C.; Buist, R.; Johnston, J.B.; Del Bigio, M.R.; Ni, W.; Dawood, M.R.; Peeling, J. Neurovirulence in feline immunodeficiency virus-infected neonatal cats is viral strain specific and dependent on systemic immune suppression. J. Virol. 1998, 72, 9109–9115. [Google Scholar] [PubMed]
- Fletcher, N.F.; Meeker, R.B.; Hudson, L.C.; Callanan, J.J. The neuropathogenesis of feline immunodeficiency virus infection: Barriers to overcome. Vet. J. 2011, 188, 260–269. [Google Scholar] [CrossRef] [PubMed]
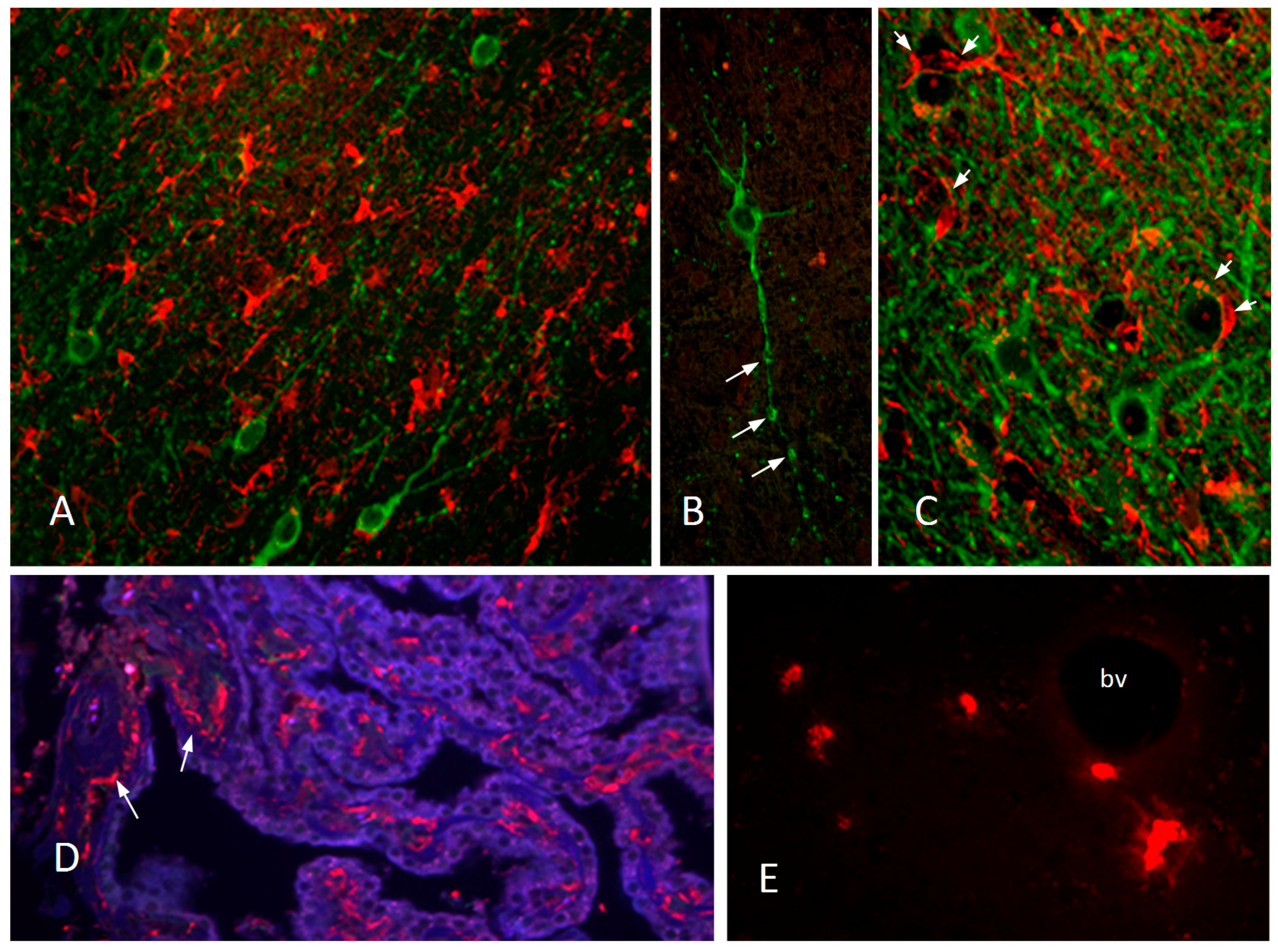
- Hudson, L.C.; Bragg, D.C.; Tompkins, M.B.; Meeker, R.B. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005, 1058, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.F.; Bexiga, M.G.; Brayden, D.J.; Brankin, B.; Willett, B.J.; Hosie, M.J.; Jacque, J.M.; Callanan, J.J. Lymphocyte migration through the blood-brain barrier (BBB) in feline immunodeficiency virus infection is significantly influenced by the pre-existence of virus and tumour necrosis factor (TNF)-alpha within the central nervous system (CNS): Studies using an in vitro feline bbb model. Neuropathol. Appl. Neurobiol. 2009, 35, 592–602. [Google Scholar] [PubMed]
- Persidsky, Y.; Stins, M.; Way, D.; Witte, M.H.; Weinand, M.; Kim, K.S.; Bock, P.; Gendelman, H.E.; Fiala, M. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J. Immunol. 1997, 158, 3499–3510. [Google Scholar] [PubMed]
- Ryan, G.; Grimes, T.; Brankin, B.; Mabruk, M.J.; Hosie, M.J.; Jarrett, O.; Callanan, J.J. Neuropathology associated with feline immunodeficiency virus infection highlights prominent lymphocyte trafficking through both the blood-brain and blood-choroid plexus barriers. J. Neurovirol. 2005, 11, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Pistello, M.; Carli, M.A.; Abramo, F.; Mancuso, G.; Nicoletti, E.; Bendinelli, M. Tumor necrosis factor-alpha and virus expression in the central nervous system of cats infected with feline immunodeficiency virus. J. Neurovirol. 1999, 5, 465–473. [Google Scholar] [CrossRef] [PubMed]
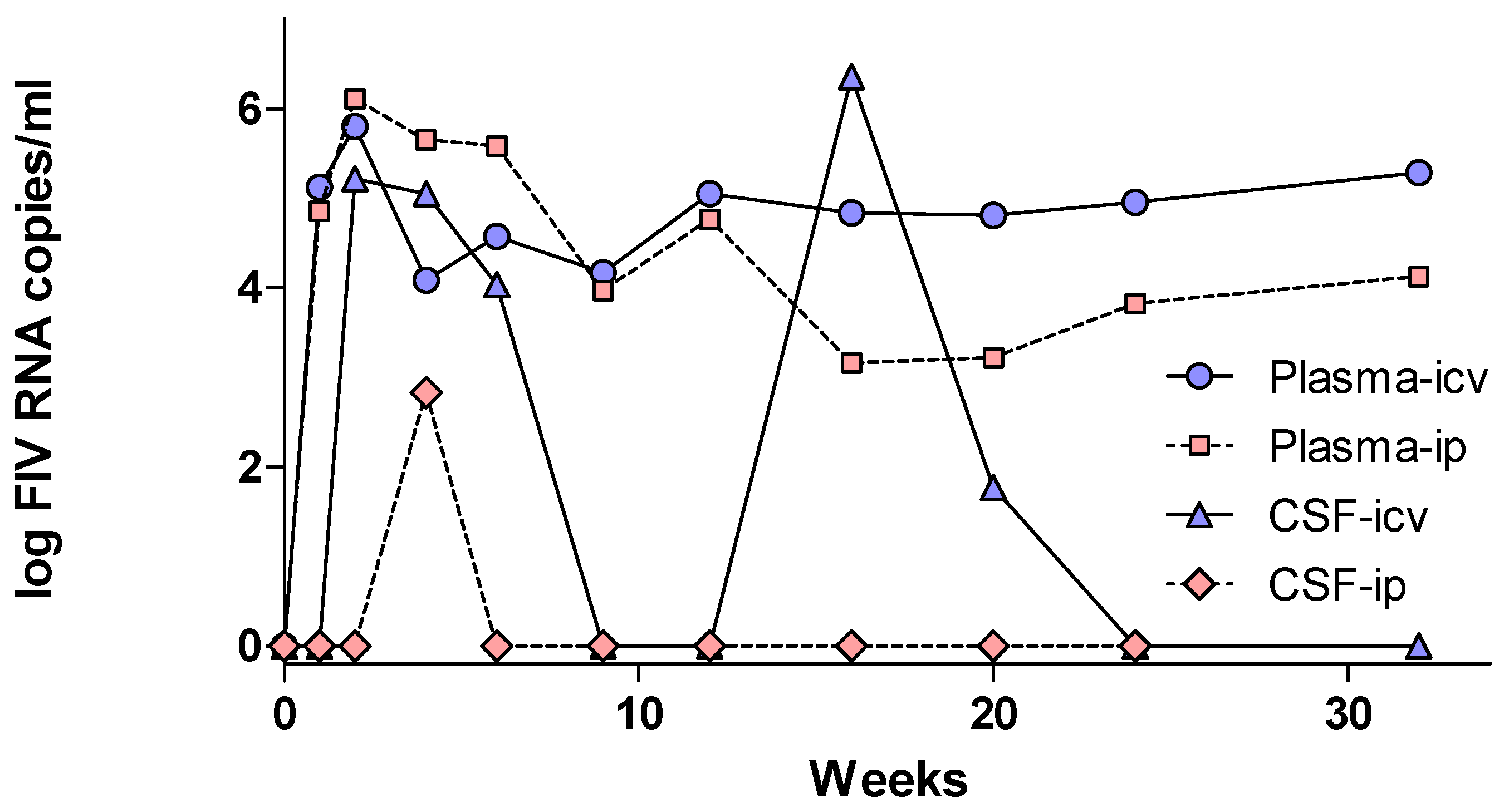
- Ryan, G.; Klein, D.; Knapp, E.; Hosie, M.J.; Grimes, T.; Mabruk, M.J.; Jarrett, O.; Callanan, J.J. Dynamics of viral and proviral loads of feline immunodeficiency virus within the feline central nervous system during the acute phase following intravenous infection. J. Virol. 2003, 77, 7477–7485. [Google Scholar] [CrossRef] [PubMed]
- Pistello, M.; Menzo, S.; Giorgi, M.; Da Prato, L.; Cammarota, G.; Clementi, M.; Bendinelli, M. Competitive polymerase chain reaction for quantitating feline immunodeficiency virus load in infected cat tissues. Mol. Cell. Probes 1994, 8, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Bragg, D.C.; Poulton, W.; Hudson, L. Transmigration of macrophages across the choroid plexus epithelium in response to the feline immunodeficiency virus. Cell Tissue Res. 2012, 347, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Steffen, B.J.; Breier, G.; Butcher, E.C.; Schulz, M.; Engelhardt, B. Icam-1, vcam-1, and madcam-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am. J. Pathol. 1996, 148, 1819–1838. [Google Scholar] [PubMed]
- Falangola, M.F.; Hanly, A.; Galvao-Castro, B.; Petito, C.K. Hiv infection of human choroid plexus: A possible mechanism of viral entry into the cns. J. Neuropathol. Exp. Neurol. 1995, 54, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Hanly, A.; Petito, C.K. Hla-dr-positive dendritic cells of the normal human choroid plexus: A potential reservoir of HIV in the central nervous system. Hum. Pathol. 1998, 29, 88–93. [Google Scholar] [CrossRef]
- Petito, C.K.; Chen, H.; Mastri, A.R.; Torres-Munoz, J.; Roberts, B.; Wood, C. Hiv infection of choroid plexus in aids and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection. J. Neurovirol. 1999, 5, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Ritola, K.; Robertson, K.; Fiscus, S.A.; Hall, C.; Swanstrom, R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of hiv-1-associated dementia. J. Virol. 2005, 79, 10830–10834. [Google Scholar] [CrossRef] [PubMed]
- Schnell, G.; Spudich, S.; Harrington, P.; Price, R.W.; Swanstrom, R. Compartmentalized human immunodeficiency virus type 1 originates from long-lived cells in some subjects with HIV-1-associated dementia. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Sturdevant, C.B.; Joseph, S.B.; Schnell, G.; Price, R.W.; Swanstrom, R.; Spudich, S. Compartmentalized replication of r5 t cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Burkala, E.J.; He, J.; West, J.T.; Wood, C.; Petito, C.K. Compartmentalization of HIV-1 in the central nervous system: Role of the choroid plexus. AIDS 2005, 19, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Strain, M.C.; Letendre, S.; Pillai, S.K.; Russell, T.; Ignacio, C.C.; Gunthard, H.F.; Good, B.; Smith, D.M.; Wolinsky, S.M.; Furtado, M.; et al. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J. Virol. 2005, 79, 1772–1788. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wood, C.; Petito, C.K. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: Potential role of choroid plexus in the pathogenesis of hiv encephalitis. J. Neurovirol. 2000, 6, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Gamst, A.C.; Capparelli, E.; Spector, S.A.; Hsia, K.; Wolfson, T.; Abramson, I.; Grant, I.; McCutchan, J.A. Cerebrospinal fluid hiv rna originates from both local cns and systemic sources. Neurology 2000, 54, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Shechter, R.; Miller, O.; Yovel, G.; Rosenzweig, N.; London, A.; Ruckh, J.; Kim, K.W.; Klein, E.; Kalchenko, V.; Bendel, P.; et al. Recruitment of beneficial m2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 2013, 38, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Prospero-Garcia, O.; Puaoi, D.; Lerner, D.; Fox, H.; Olmsted, R.; Bloom, F.; Heriksen, S.; Elder, J. Neurological abnormalities associated with feline immunodeficiency virus infection. J. Gen. Virol. 1994, 75, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.R.; Prospero-Garcia, O.; Wheeler, D.W.; Wagaman, P.C.; Lerner, D.L.; Fox, H.S.; Whalen, L.R.; Bloom, F.E.; Elder, J.H.; Henriksen, S.J. Neurologic dysfunctions caused by a molecular clone of feline immunodeficiency virus, FIV-ppr. J. Neurovirol. 1996, 2, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Hayes, K.; Oglesbee, M.; Mathes, L. Progressive encephalopathy associated with cd4/cd8 inversion in adult FIV-infected cats. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Maruyama, K.; Smith, M.; Hayes, K.A.; Buck, W.R.; Ruehlmann, D.S.; Mathes, L.E. Frontal lobe neuronal injury correlates to altered function in FIV-infected cats. J. Acquir. Immune Defic. Syndr. 1999, 22, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Oglesbee, M.; Mathes, L.; Krakowka, S.; Olmstead, R.; Lafrado, L. Aids-associated encephalopathy with experimental feline immunodeficiency virus infection. J. Acquir. Immune Defic. Syndr. 1993, 6, 758–771. [Google Scholar] [PubMed]
- Henriksen, S.J.; Prospero-Garcia, O.; Phillips, T.R.; Fox, H.S.; Bloom, F.E.; Elder, J.H. Feline immunodeficiency virus as a model for study of lentivirus infection of the central nervous system. Curr. Top. Microbiol. Immunol. 1995, 202, 167–186. [Google Scholar] [PubMed]
- Buck, W.R.; Podell, M. Neuronal loss in fiv-md infected cats. J. NeuroAIDS. 1998, 2, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.J.; Hayes, K.A.; Buck, W.R.; Podell, M.; Mathes, L.E. Neurophysiologic and immunologic abnormalities associated with feline immunodeficiency virus molecular clone FIV-ppr DNA inoculation. J. Acquir. Immune Defic. Syndr. 2000, 23, 8–16. [Google Scholar] [CrossRef] [PubMed]
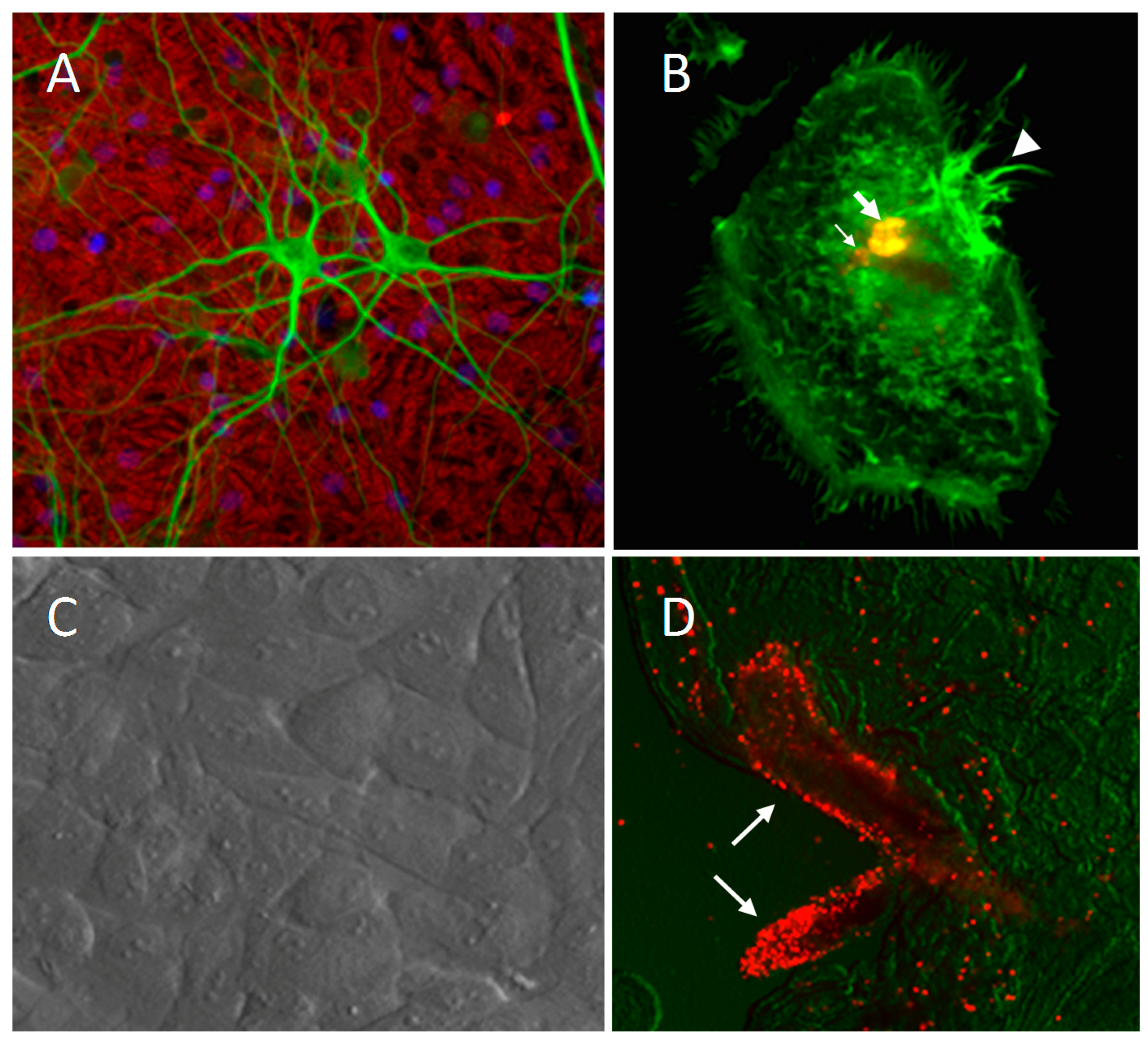
- Bragg, D.C.; Hudson, L.C.; Liang, Y.H.; Tompkins, M.B.; Fernandes, A.; Meeker, R.B. Choroid plexus macrophages proliferate and release toxic factors in response to feline immunodeficiency virus. J. NeuroVirol. 2002, 8, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Abramo, F.; Bo, S.; Canese, M.G.; Poli, A. Regional distribution of lesions in the central nervous system of cats infected with feline immunodeficiency virus. AIDS Res. Hum. Retrovir. 1995, 11, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Hurtrel, M.; Gray, F.; Claessens-Maire, M.A.; Ganiere, J.P.; Montagnier, L.; Hurtrel, B. Virus load and neuropathology in the FIV model. J. NeuroVirol. 1996, 2, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Hurtrel, M.; Ganiere, J.; Guelifi, J.; Chakrabarti, L.; Maire, M.; Gray, F.; Montagnier, L.; Hurtrel, B. Comparison of early and late feline immunodeficiency virus encephalopathies. AIDS 1992, 6, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Abramo, F.; Di Iorio, C.; Cantile, C.; Carli, M.A.; Pollera, C.; Vago, L.; Tosoni, A.; Costanzi, G. Neuropathology in cats experimentally infected with feline immunodeficiency virus: A morphological, immunocytochemical and morphometric study. J. Neurovirol. 1997, 3, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Silvotti, L.; Corradi, A.; Brandi, G.; Cabassi, A.; Bendinelli, M.; Magnan, M.; Piedimonte, G. FIV induced encephalopathy: Early brain lesions in the absence of viral replication in monocyte/macrophages. A pathogenetic model. Vet. Immunol. Immunopathol. 1997, 55, 263–271. [Google Scholar] [CrossRef]
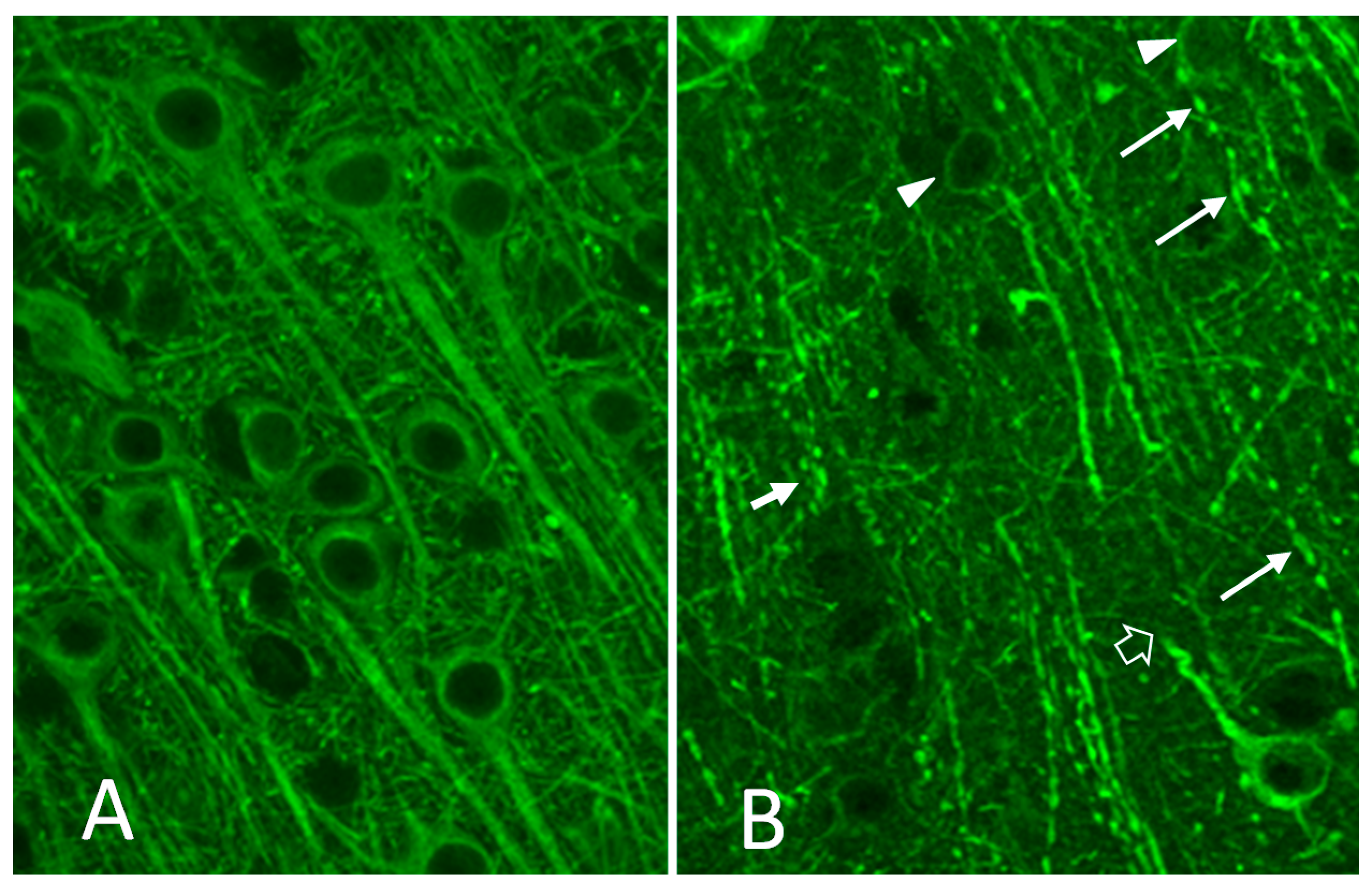
- Jacobson, S.; Henriksen, S.J.; Prospero-Garcia, O.; Phillips, T.R.; Elder, J.H.; Young, W.G.; Bloom, F.E.; Fox, H.S. Cortical neuronal cytoskeletal changes associated with FIV infection. J. Neurovirol. 1997, 3, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Koirala, T.R.; Nakagaki, K.; Ishida, T.; Nonaka, S.; Morikawa, S.; Tabira, T. Decreased expression of map-2 and gad in the brain of cats infected with feline immunodeficiency virus. Tohoku J. Exp. Med. 2001, 195, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Thiede, B.A.; Hall, C.; English, R.; Tompkins, M. Cortical cell loss in asymptomatic cats experimentally infected with feline immunodeficiency virus. AIDS Res. Hum. Retrovir. 1997, 13, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.W.; Buckmaster, P.S.; Hoover, E.A.; Whalen, L.R.; Dudek, F.E. Neuron loss and axon reorganization in the dentate gyrus of cats infected with the feline immunodeficiency virus. J. Comp. Neurol. 1999, 411, 563–577. [Google Scholar] [CrossRef]
- Head, E.; Moffat, K.; Das, P.; Sarsoza, F.; Poon, W.W.; Landsberg, G.; Cotman, C.W.; Murphy, M.P. Beta-amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiol. Aging 2005, 26, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Gunn-Moore, D.A.; McVee, J.; Bradshaw, J.M.; Pearson, G.R.; Head, E.; Gunn-Moore, F.J. Ageing changes in cat brains demonstrated by beta-amyloid and at8-immunoreactive phosphorylated tau deposits. J. Feline Med. Surg. 2006, 8, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.K.; Tokuda, T.; Uchida, K.; Ishii, R.; Tatebe, H.; Takahashi, E.; Tomiyama, T.; Une, Y.; Nakayama, H. The domestic cat as a natural animal model of alzheimer’s disease. Acta Neuropathol. Commun. 2015, 3, 78. [Google Scholar] [CrossRef] [PubMed]
- Asproni, P.; Abramo, F.; Millanta, F.; Lorenzi, D.; Poli, A. Amyloidosis in association with spontaneous feline immunodeficiency virus infection. J. Feline Med. Surg. 2013, 15, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, G.M.; Denenberg, S.; Araujo, J.A. Cognitive dysfunction in cats: A syndrome we used to dismiss as “old age”. J. Feline Med. Surg. 2010, 12, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Gunn-Moore, D.A. Cognitive dysfunction in cats: Clinical assessment and management. Top. Companion Anim. Med. 2011, 26, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.C.; Boles, J.C.; Meeker, R.B. Destabilization of neuronal calcium homeostasis by factors secreted from choroid plexus macrophage cultures in response to feline immunodeficiency virus. Neurobiol. Dis. 2002, 9, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.C.; Meeker, R.B.; Duff, B.A.; English, R.V.; Tompkins, M.B. Neurotoxicity of FIV and fiv envelope protein in feline cortical cultures. Brain Res. 1999, 816, 431–437. [Google Scholar] [CrossRef]
- Meeker, R.; English, R.; Tompkins, M. Enhanced excitotoxicity in primary feline neural cultures exposed to feline immunodeficiency virus (FIV). J. Neuro-AIDS 1997, 1, 1–27. [Google Scholar] [CrossRef]
- Meeker, R.B.; Poulton, W.; Feng, W.H.; Hudson, L.; Longo, F.M. Suppression of immunodeficiency virus-associated neural damage by the p75 neurotrophin receptor ligand, lm11a-31, in an in vitro feline model. J. Neuroimmune Pharmacol. 2012, 7, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Azuma, Y.; Bragg, D.C.; English, R.V.; Tompkins, M. Microglial proliferation in cortical neural cultures exposed to feline immunodeficiency virus. J. Neuroimmunol. 1999, 101, 15–26. [Google Scholar] [CrossRef]
- Al Ghoul, W.M.; Meeker, R.B.; Greenwood, R.S. Kindling induces a long-lasting increase in brain nitric oxide synthase activity. NeuroRep. 1995, 6, 457–460. [Google Scholar] [CrossRef]
- Hudson, L.C.; Tompkins, M.B.; Meeker, R.B. Endothelial cell suppression of peripheral blood mononuclear cell trafficking in vitro during acute exposure to feline immunodeficiency virus. Cell Tissue Res. 2008, 334, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Power, C. Regulation of neural cell survival by HIV-1 infection. Neurobiol. Dis. 2006, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Avdoshina, V.; Bachis, A.; Mocchetti, I. Synaptic dysfunction in human immunodeficiency virus type-1-positive subjects: Inflammation or impaired neuronal plasticity? J. Intern. Med. 2013, 273, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.; Khan, M.Z.; Kolson, D.L. Current understanding of hiv-associated neurocognitive disorders pathogenesis. Curr. Opin. Neurol. 2011, 24, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kolson, D.L. Neuropathogenesis of central nervous system HIV-1 infection. Clin. Lab. Med. 2002, 22, 703–717. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Gelbard, H.A. Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox. Res. 2005, 8, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Turchan, J.; Sacktor, N.; Wojna, V.; Conant, K.; Nath, A. Neuroprotective therapy for hiv dementia. Curr. HIV Res. 2003, 1, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Boles, J.C.; Robertson, K.R.; Hall, C.D. Cerebrospinal fluid from human immunodeficiency virus—Infected individuals facilitates neurotoxicity by suppressing intracellular calcium recovery. J. NeuroVirol. 2005, 11, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.G.; Gelbard, H.A. HIV-1-induced neuronal injury in the developing brain. J. Leukoc. Biol. 1999, 65, 453–457. [Google Scholar] [PubMed]
- Gemignani, A.; Paudice, P.; Pittaluga, A.; Raiteri, M. The HIV-1 coat protein gp120 and some of its fragments potently activate native cerebral nmda receptors mediating neuropeptide release. Eur. J. Neurosci. 2000, 12, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Nath, A.; Mattson, M.P.; Slevin, J.T.; Geiger, J.D. HIV-1 tat through phosphorylation of nmda receptors potentiates glutamate excitotoxicity. J. Neurochem. 2001, 78, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.; Sucher, N.; Kaiser, P.; Dreyer, E. Synergistic effects of hiv coat protein and nmda receptor-mediated neurotoxicity. Neuron 1991, 7, 111–118. [Google Scholar] [CrossRef]
- Lipton, S. Models of neuronal injury in aids: Another role for the nmda receptor? TINS 1992, 15, 75–80. [Google Scholar] [CrossRef]
- Self, R.L.; Mulholland, P.J.; Nath, A.; Harris, B.R.; Prendergast, M.A. The human immunodeficiency virus type-1 transcription factor tat produces elevations in intracellular Ca2+ that require function of an n-methyl-d-aspartate receptor polyamine-sensitive site. Brain Res. 2004, 995, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Fine, S.M.; Angel, R.A.; Perry, S.W.; Epstein, L.G.; Rothstein, J.D.; Dewhurst, S.; Gelbard, H.A. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J. Biol. Chem. 1996, 271, 15303–15306. [Google Scholar] [PubMed]

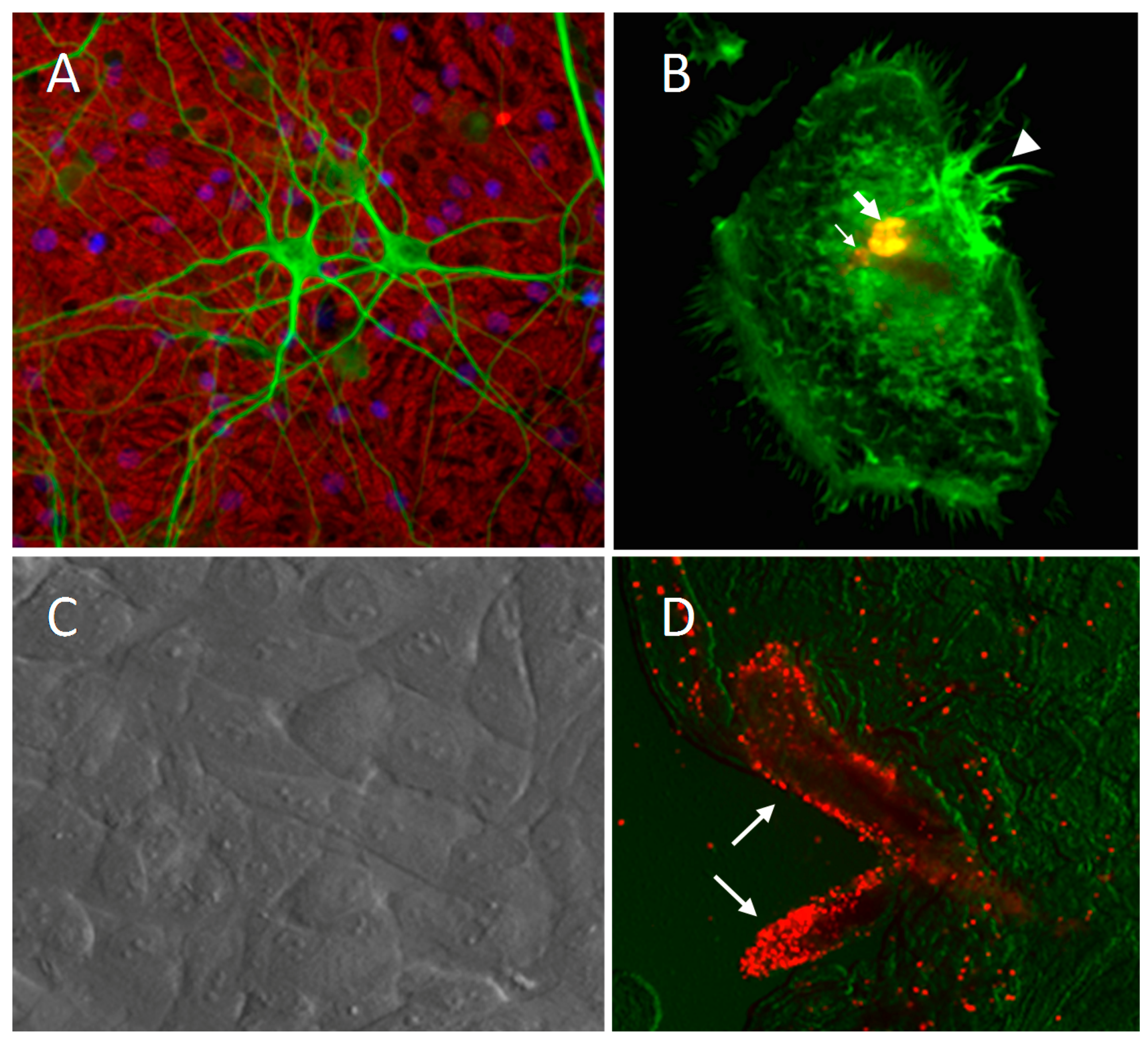
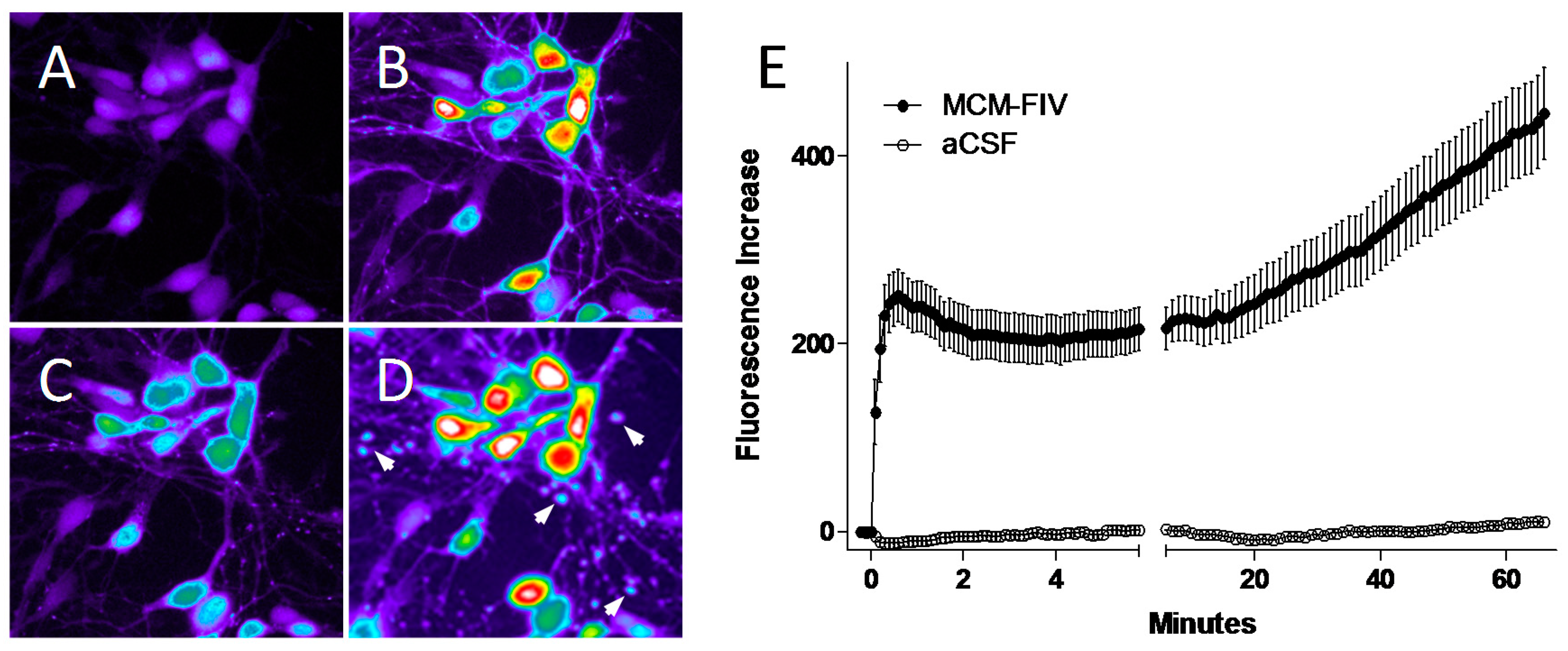
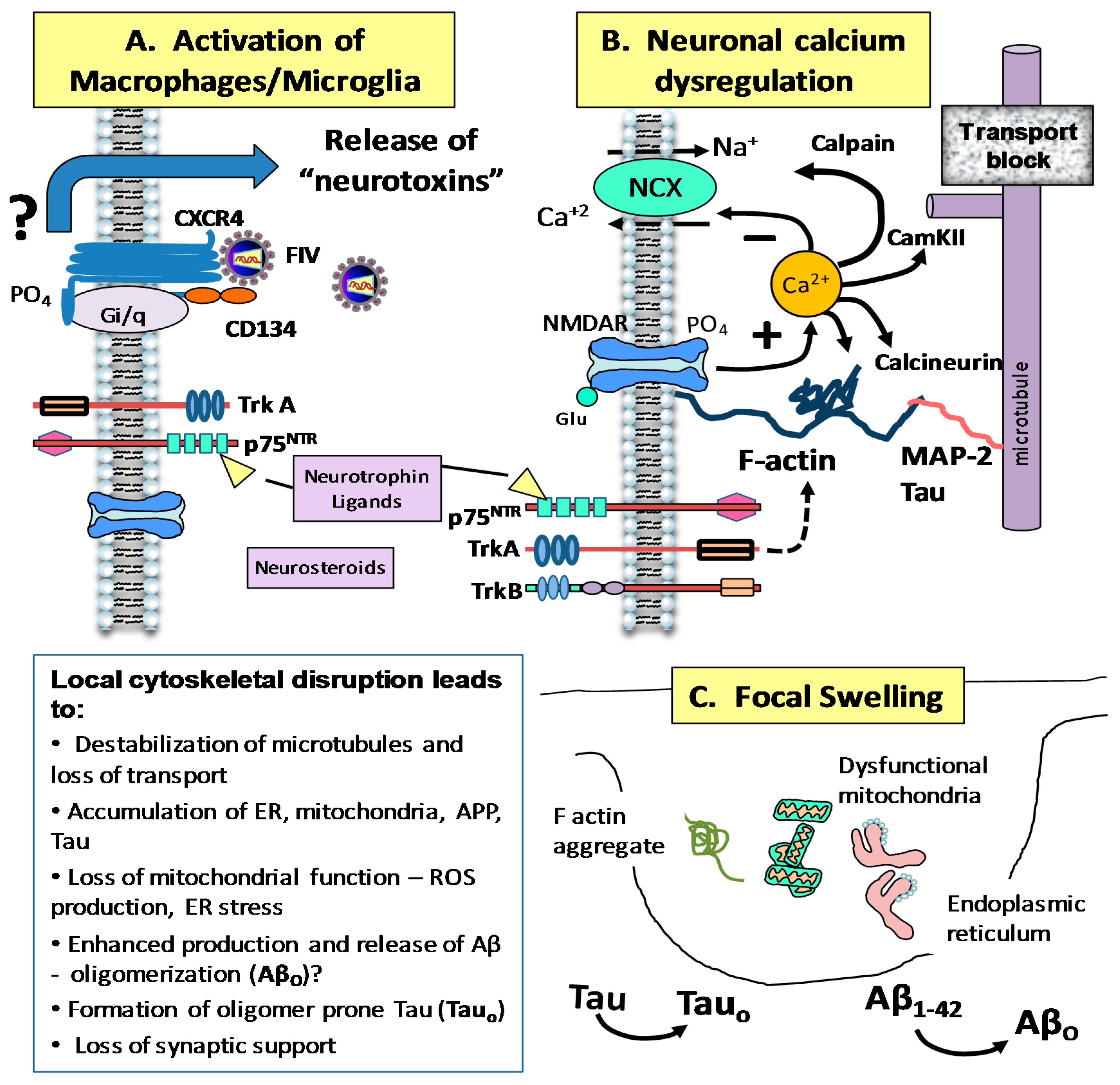
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meeker, R.B.; Hudson, L. Feline Immunodeficiency Virus Neuropathogenesis: A Model for HIV-Induced CNS Inflammation and Neurodegeneration. Vet. Sci. 2017, 4, 14. https://doi.org/10.3390/vetsci4010014
Meeker RB, Hudson L. Feline Immunodeficiency Virus Neuropathogenesis: A Model for HIV-Induced CNS Inflammation and Neurodegeneration. Veterinary Sciences. 2017; 4(1):14. https://doi.org/10.3390/vetsci4010014
Chicago/Turabian StyleMeeker, Rick B., and Lola Hudson. 2017. "Feline Immunodeficiency Virus Neuropathogenesis: A Model for HIV-Induced CNS Inflammation and Neurodegeneration" Veterinary Sciences 4, no. 1: 14. https://doi.org/10.3390/vetsci4010014
APA StyleMeeker, R. B., & Hudson, L. (2017). Feline Immunodeficiency Virus Neuropathogenesis: A Model for HIV-Induced CNS Inflammation and Neurodegeneration. Veterinary Sciences, 4(1), 14. https://doi.org/10.3390/vetsci4010014