
Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
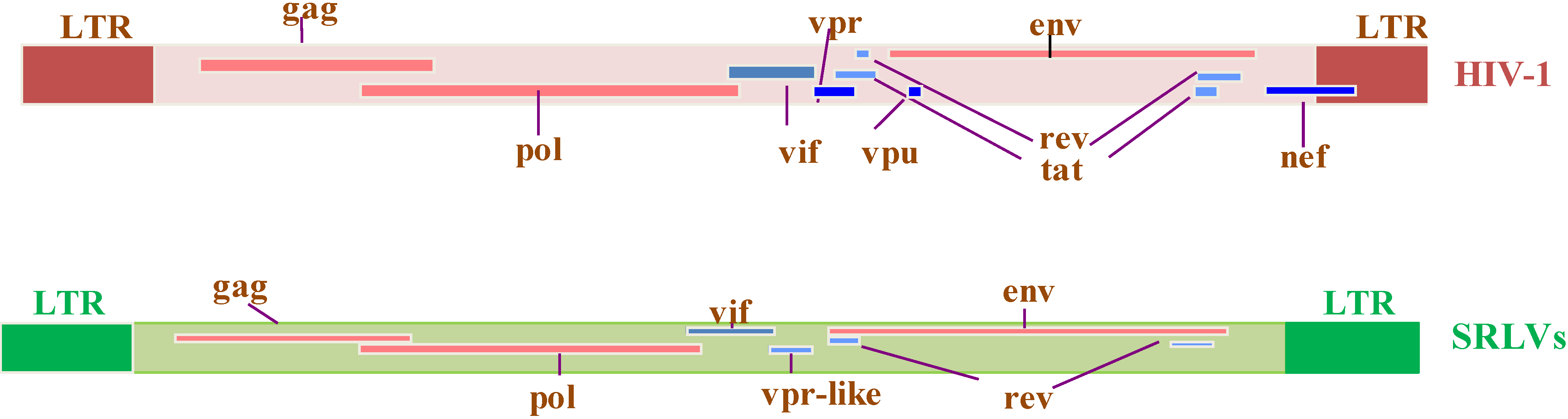
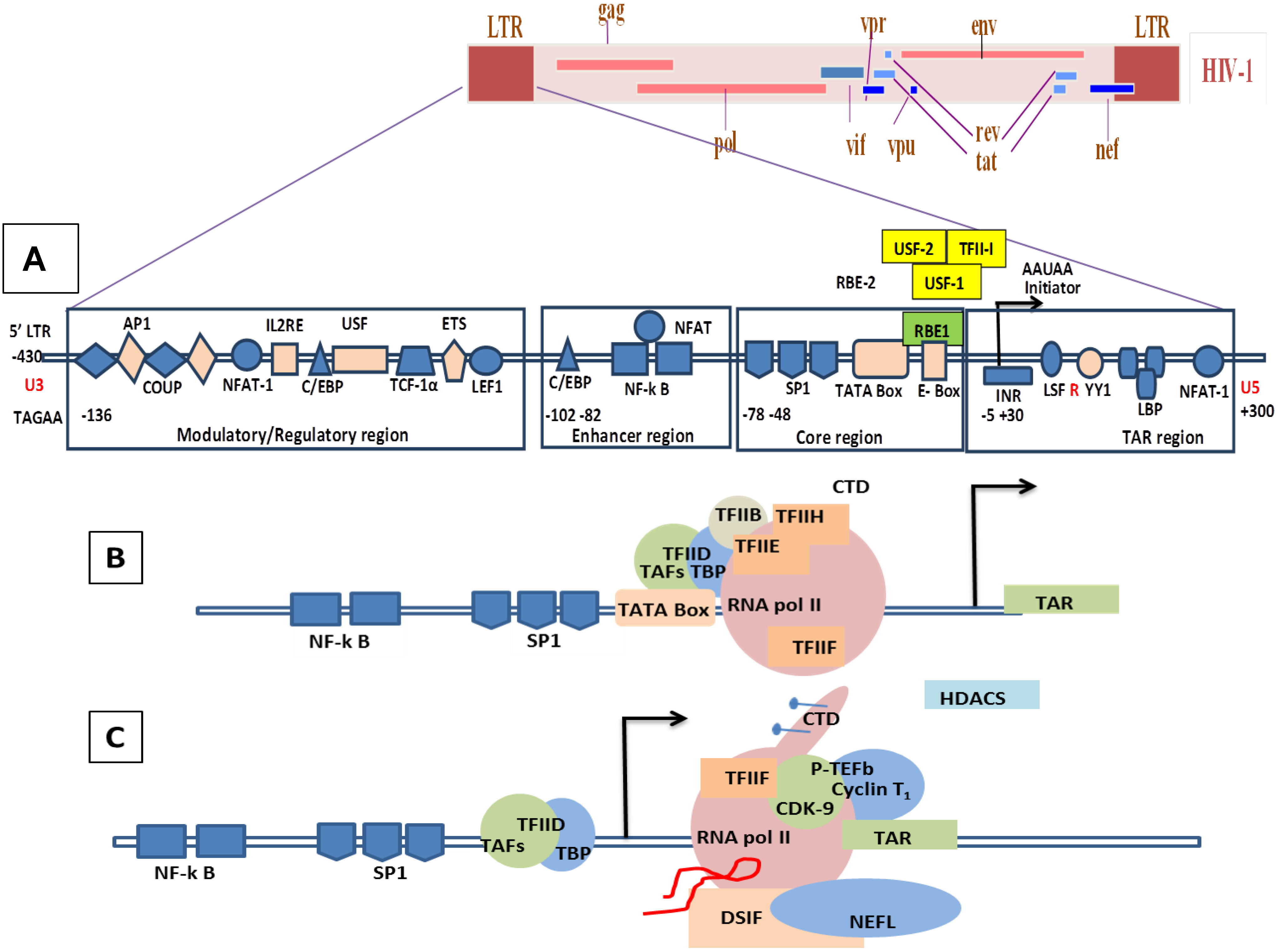
2. Genome Organization of SRLVs and HIV-1
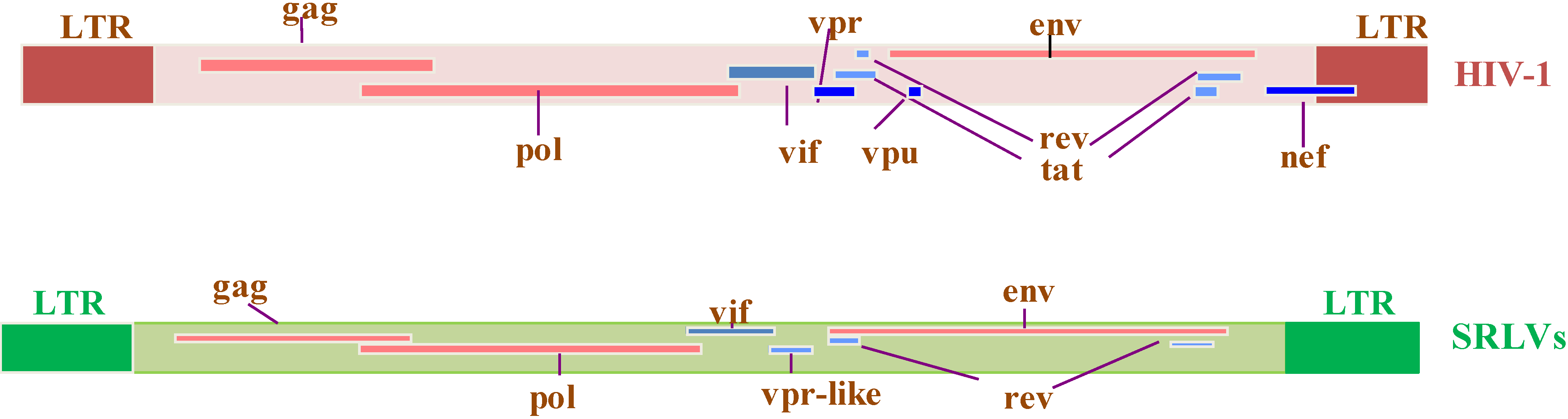
2.1. Virus-Encoded Regulatory Proteins
2.1.1. Regulatory Tat
2.1.2. Regulatory Rev
2.1.3. Regulatory Vif
2.2. Accessory Virus Gene Encoded Proteins
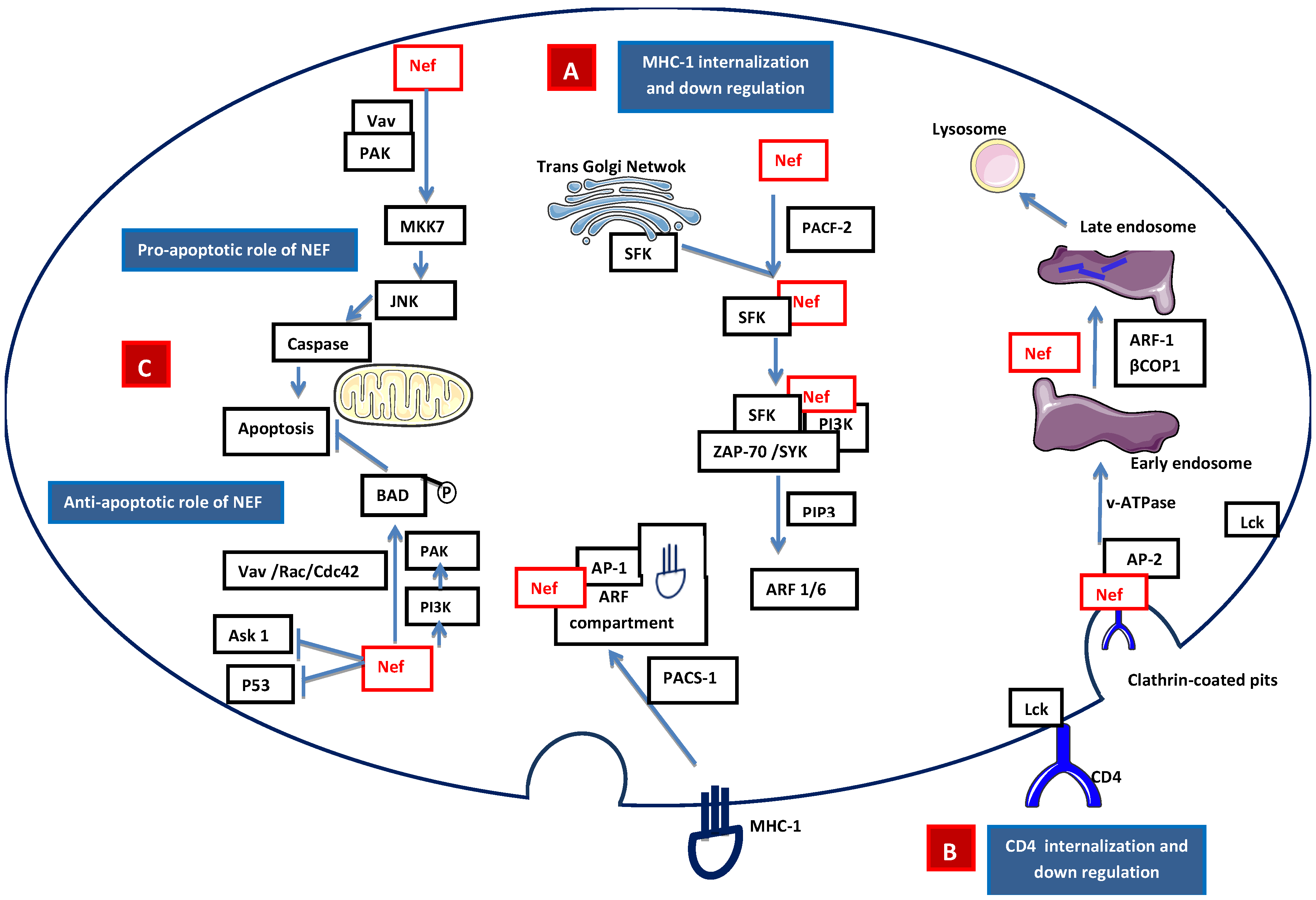
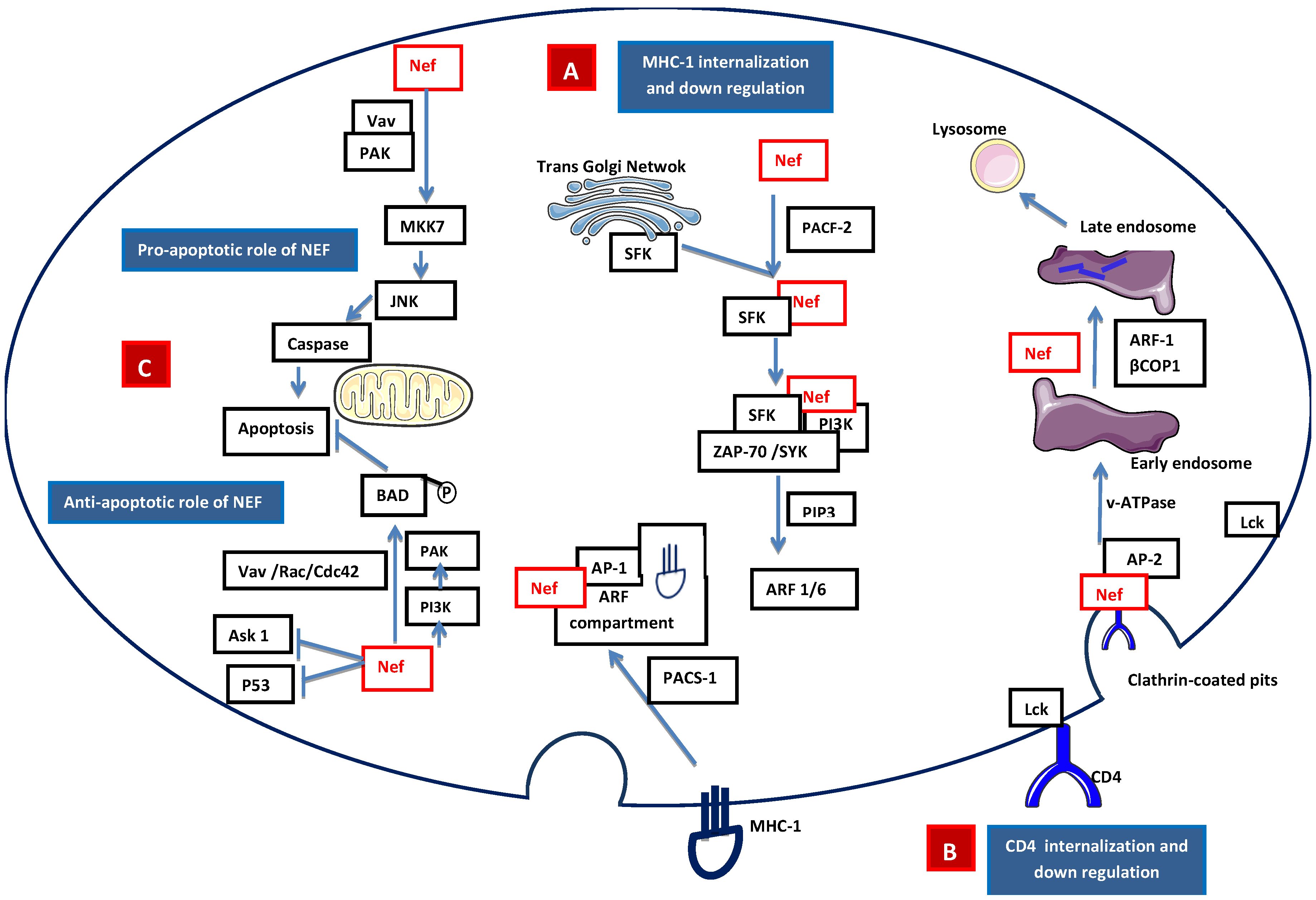
2.2.1 Accessory Nef
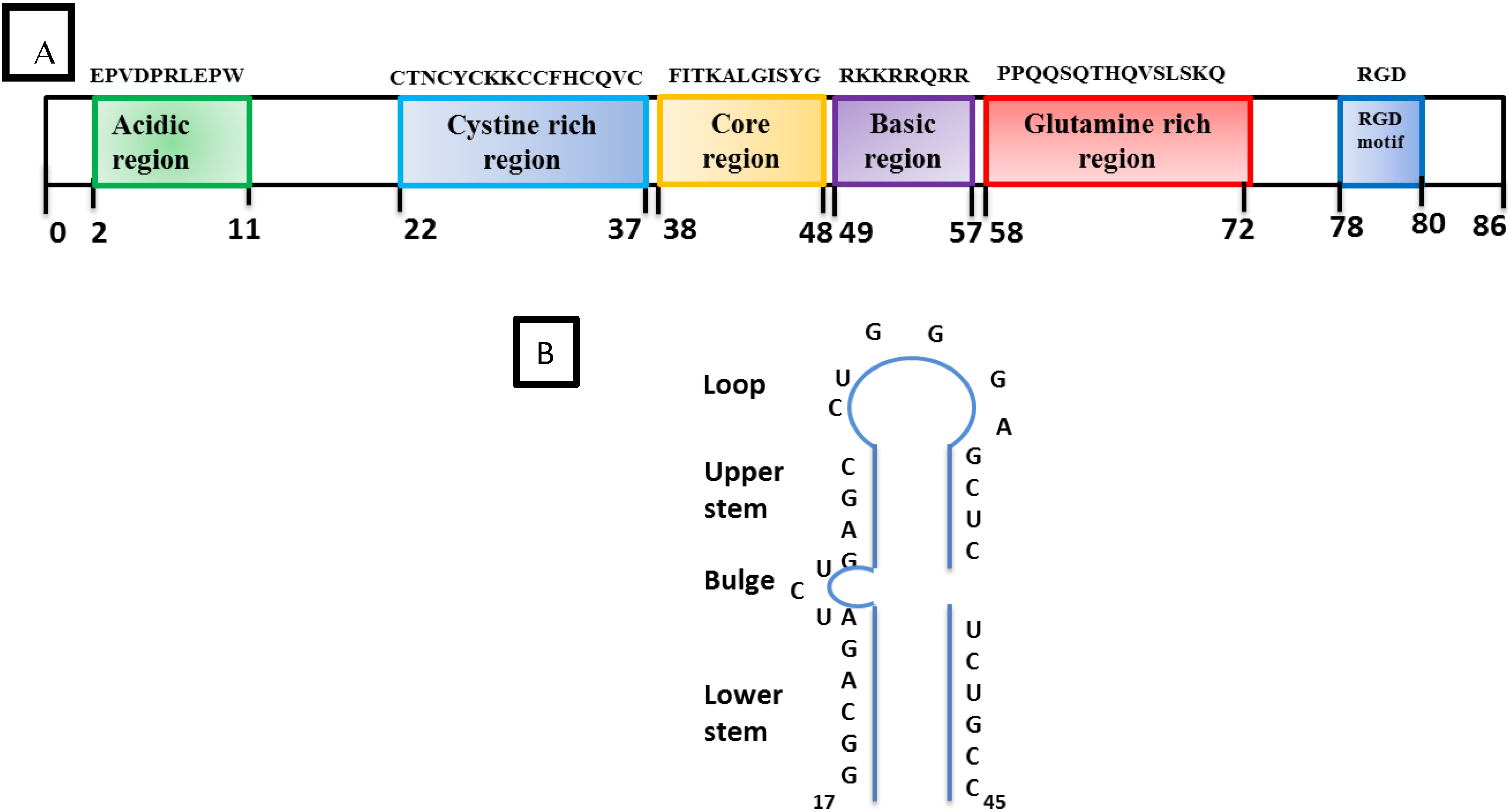
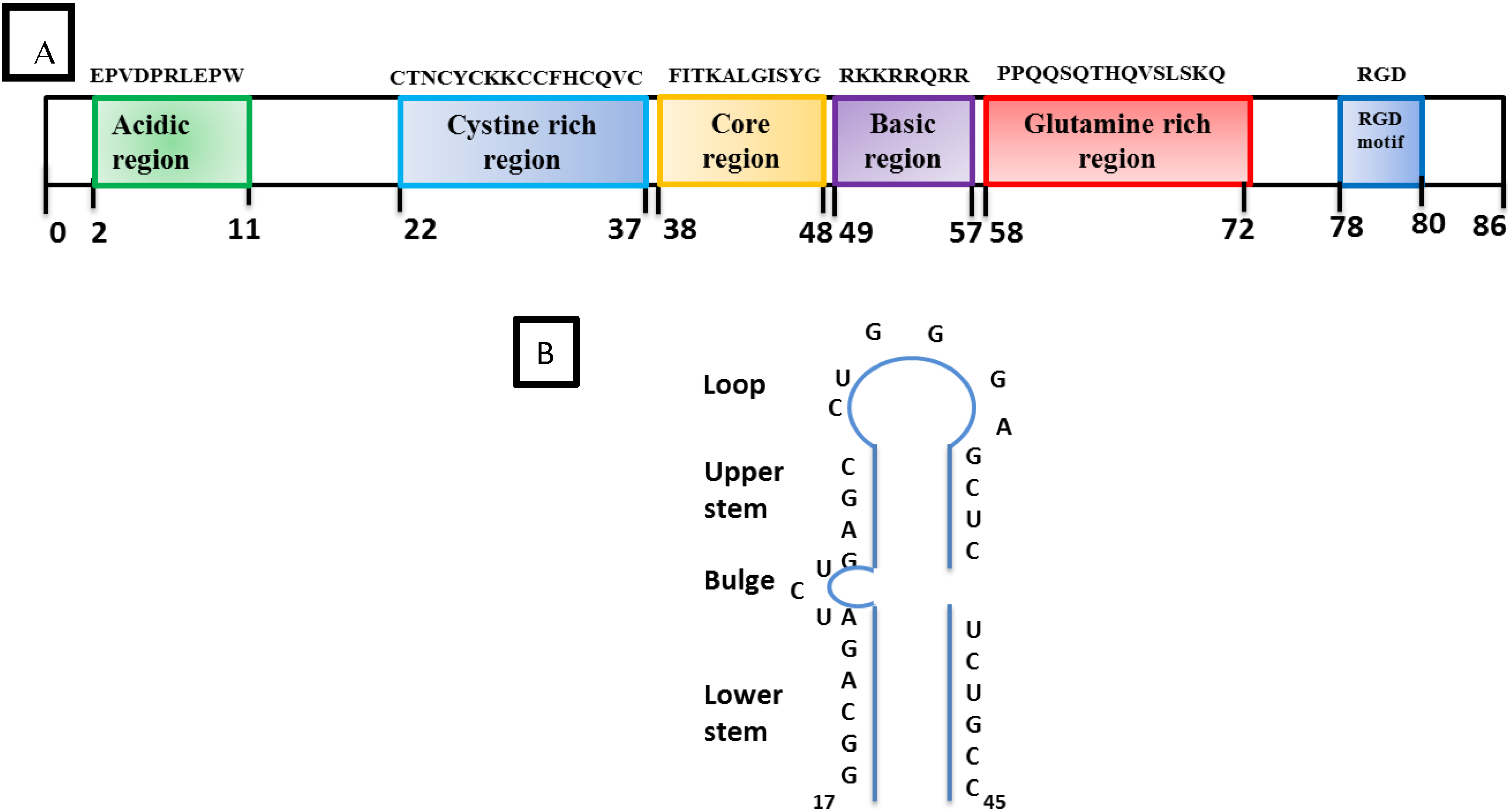
2.2.2. Accessory Vpr
2.3. HIV Tat Protein
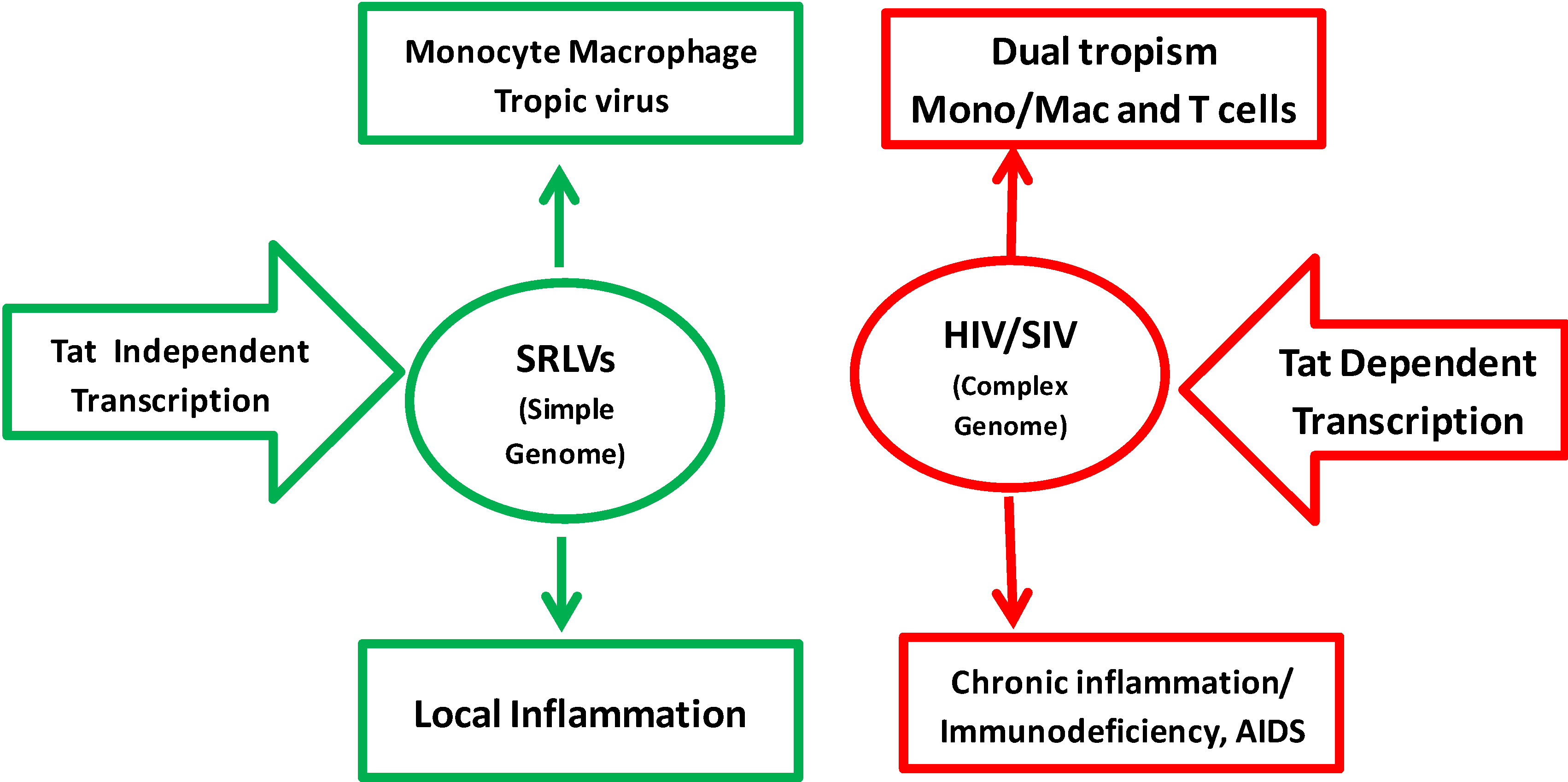
2.4. Absence of SRLV Tat
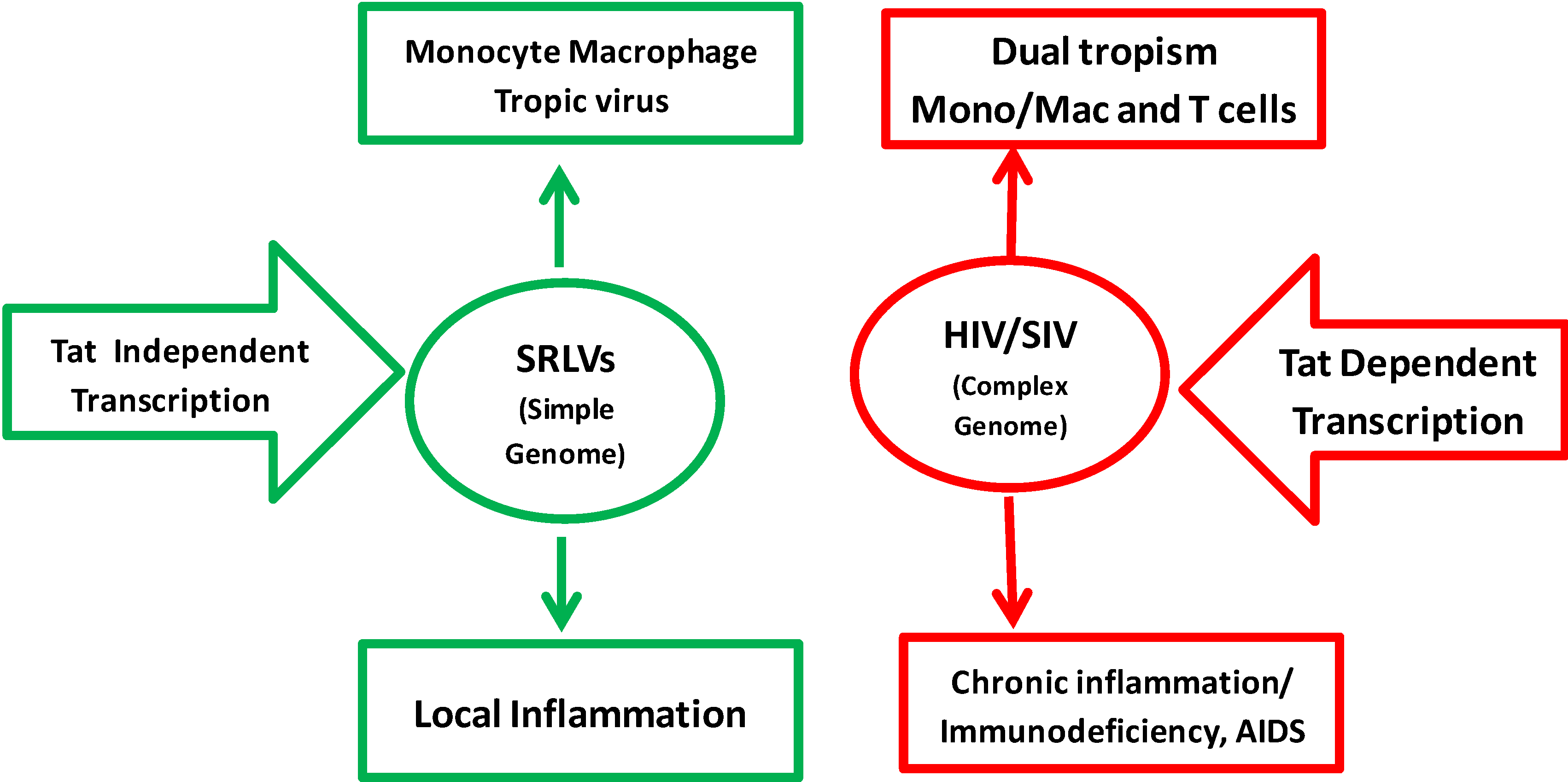
3. Natural History of HIV-1 and SRLV
3.1. Discovery of HIV-1
3.2. Cross-Species Infection from Monkeys to Human
3.3. HIV Transmission in Humans
3.4. HIV Disease Progression
3.5. First Descriptions of SRLV Infection
3.6. Cellular Tropism in Vivo and in Vitro
3.7. Cross-Species Infection
3.8. Natural SRLV Transmission
3.9. SRLV Viral Dynamics in Vivo
3.10. Acute Primary Infection, Disease Progression, and Characterization
4. Clinical Pathogenesis
5. Receptor/Co-Receptor Usage
6. HAART in HIV-Infected Patients
7. Latency and Persistence
- Absence of Tat, or non-functional transactivation activity of Tat [210].
- Epigenetic regulations/chromatin remodeling by post-transcriptional modifications (hypoacetylation or trimethylation) [211].
- Influence of integration sites and provirus orientation on HIV-1 transcription efficiency [216].
- Unproductive control of viral RNA splicing, due to absence of Rev and innate host antiviral processes [217].
8. Molecular Biology of HIV-1 Latency
8.1. Molecular Mechanisms of the Transcription Involving Tat in HIV/SIV
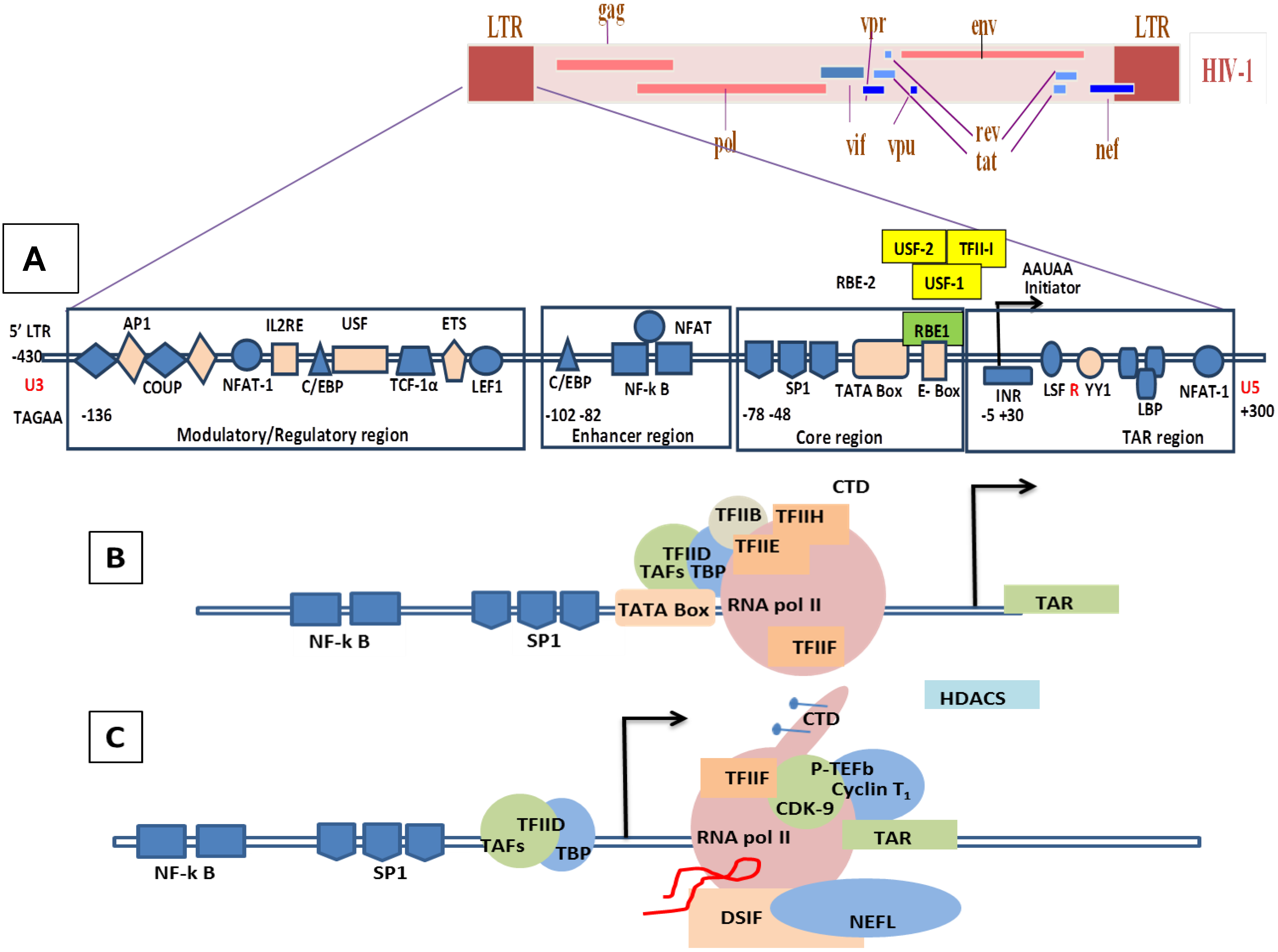
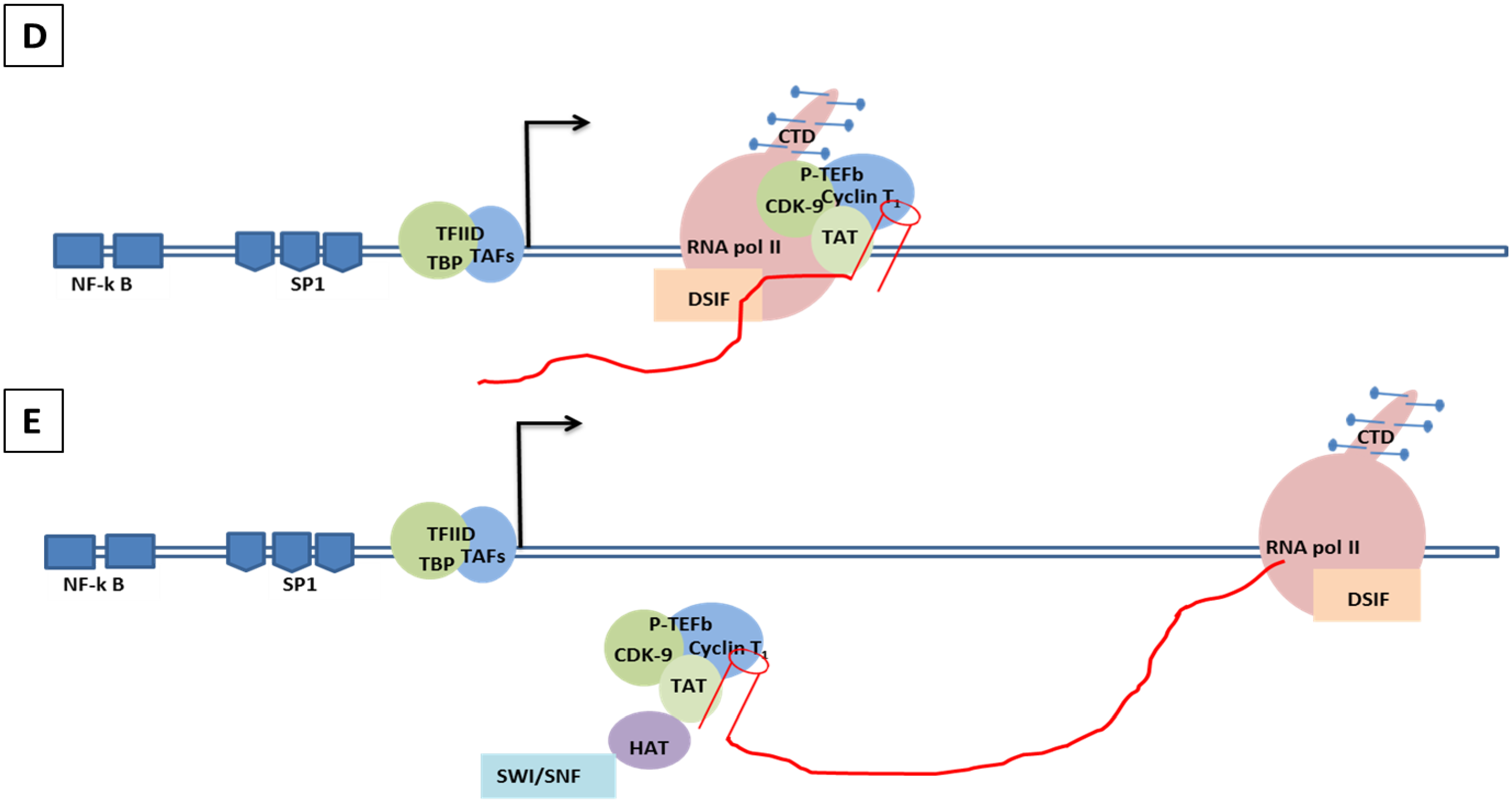
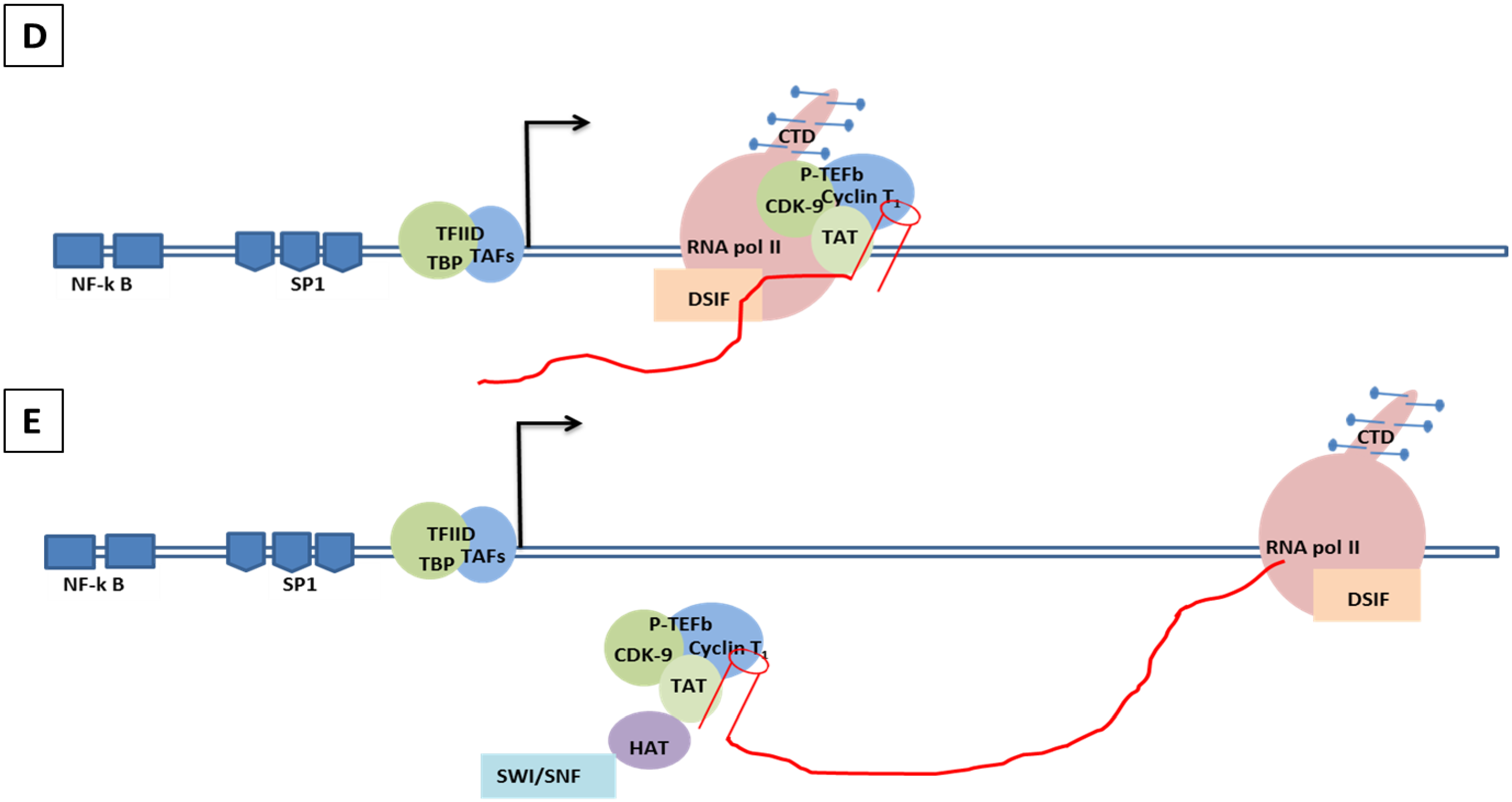
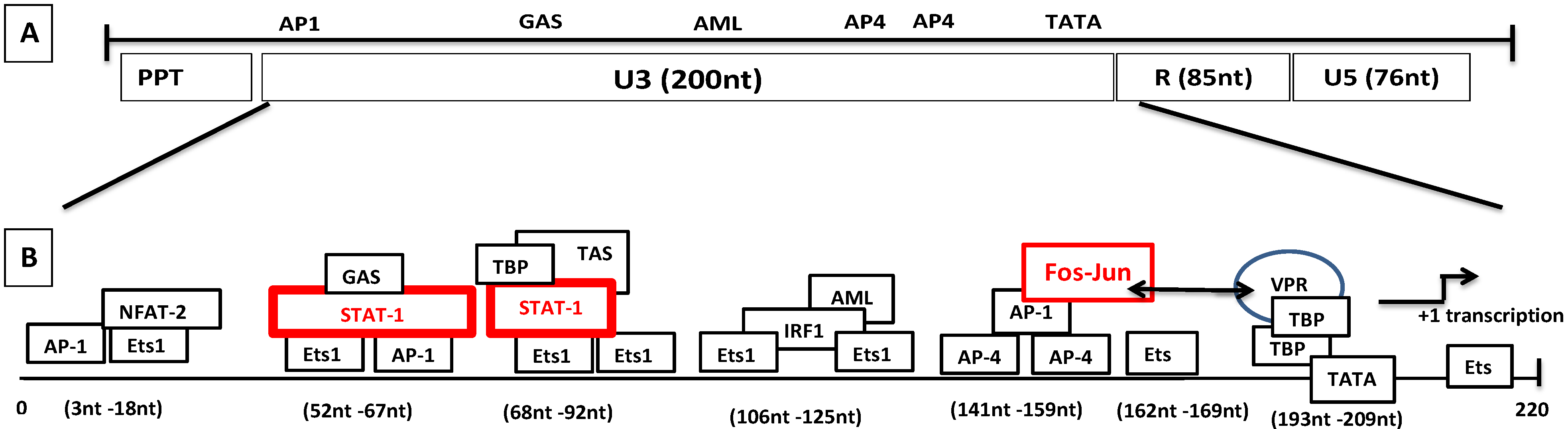
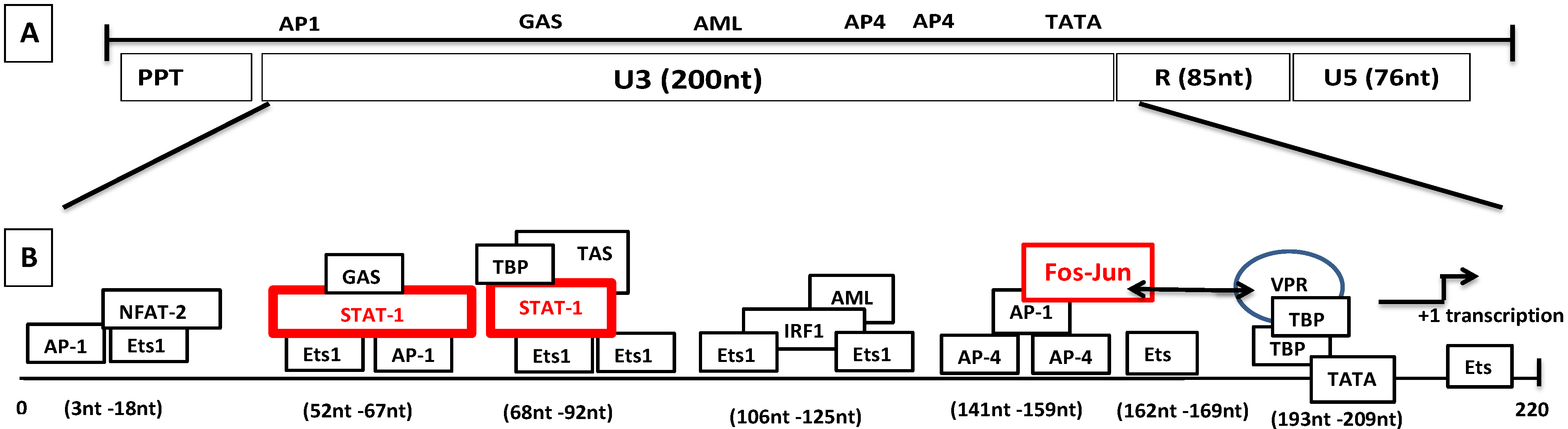
8.2. Molecular Mechanism of Tat-Independent Transcription in SRLVs
- (1).
- Vpr-like protein mediates transcription activation of SRLV genomes and this is dependent on the presence of AP-1 and AP-4 sites located in the U3 region of SRLV LTRs. The proximal AP1 site to the MVV TATA box promoter is the most important for transcription, though SRLV Vpr-like protein does not bind the AP-1 site [264,268].
- (2).
- SRLV Vpr-like protein structure has three domains including: (a) an N-terminal acidic and hydrophobic domain, presumably the activator domain, which interacts with the TATA binding protein (TBP) in vitro [59,266]; (b) a central Leucine-rich domain that can interact with FOS-Jun-specific proteins in the AP-1 sites [266]; (c) a C-terminal Cysteine-rich domain which might direct protein dimerization. An interaction of the SRLV Vpr-like protein with the TBP from the TFIID complex was shown to bind to the TATA box via its activator domain, in addition to the proximal AP-1 binding to the Leucine-rich domain. These interactions allow a stabilization of protein complexes located at the viral promoter, and therefore induce an increase of viral gene transcription as illustrated in Figure 6.
8.3. Role of Epigenetics and Transcriptional Regulation Factors in Modulating HIV-1 Latency
8.4. Latency and HIV Cure
- Reactivation of latent virus while patient is on HAART using HDAC inhibitors, vorinostats (SAHA), valproic acid, [317] “non-oncogenic” phorbol ester (bryostatin, prostratin), and cytokine IL-7 [318]. Induction of viral replication of proviruses in cells from latent reservoirs and clearance is the most promising “Kick and kill” strategy.
- Reversal of immune exhaustion (PD-1 antibodies) [329],
The Unique Case of Possible Cure
9. Sites of Latent Reservoirs
9.1. Cell Lineages
9.1.1. CD4+ T Lymphocytes
9.1.2. Monocytes and Macrophages
9.1.3. Dendritic Cells
9.2. Anatomical Reservoirs
9.2.1. CNS
9.2.2. Lymphoid Organs
9.2.3. Genital Tract
9.2.4. Lungs and Kidney
10. Can SRLV Help to Develop Strategies to Control HIV-1?
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peeters, M.; Fransen, K.; Delaporte, E.; Van den Haesevelde, M.; Gershy-Damet, G.M.; Kestens, L.; van der Groen, G.; Piot, P. Isolation and characterization of a new chimpanzee lentivirus (simian immunodeficiency virus isolate cpz-ant) from a wild-captured chimpanzee. AIDS 1992, 6, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Honore, C.; Huet, T.; Bedjabaga, L.; Ossari, S.; Bussi, P.; Cooper, R.W.; Delaporte, E. Isolation and partial characterization of an HIV-related virus occurring naturally in chimpanzees in Gabon. AIDS 1989, 3, 625–630. [Google Scholar] [CrossRef]
- Van Heuverswyn, F.; Li, Y.; Neel, C.; Bailes, E.; Keele, B.F.; Liu, W.; Loul, S.; Butel, C.; Liegeois, F.; Bienvenue, Y.; et al. Human immunodeficiency viruses: SIV infection in wild gorillas. Nature 2006. [Google Scholar] [CrossRef] [PubMed]
- Barre-Sinoussi, F.; Chermann, J.; Rey, F.; Nugeyre, M.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler, B.C.; Vezinet- Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Terzieva, V. Regulatory T Cells and HIV-1 Infection. Viral Immunol. 2008, 21, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, B.; Palsson, P.A. Visna of sheep; a slow, demyelinating infection. Br. J. Exp. Pathol. 1958, 39, 519–528. [Google Scholar] [PubMed]
- Sigurdsson, B.; Palsson, P.A.; Tryggvaddottir, A. Transmission experiments with maedi. J. Infect. Dis. 1953, 93, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Narayan, O.; Kennedy-Stoskopf, S.; Kennedy, P.G.; Ghotbi, Z.; Clements, J.E.; Stanley, J.; Pezeshkpour, G. Tropism of sheep lentiviruses for monocytes: susceptibility to infection and virus gene expression increase during maturation of monocytes to macrophages. J. Virol. 1986, 58, 67–74. [Google Scholar] [PubMed]
- Gorrell, M.D.; Brandon, M.R.; Sheffer, D.; Adams, R.J.; Narayan, O. Ovine lentivirus is macrophagetropic and does not replicate productively in T lymphocytes. J. Virol. 1992, 66, 2679–2688. [Google Scholar] [PubMed]
- Clements, J.E.; Zink, M.C. Molecular biology and pathogenesis of animal lentivirus infections. Clin. Microbiol. Rev. 1996, 9, 100–117. [Google Scholar] [PubMed]
- Fun, A.; Wensing, A.M.; Verheyen, J.; Nijhuis, M. Human immunodeficiency virus Gag and protease: Partners in resistance. Retrovirology 2012. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Krausslich, H.G.; Muller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef]
- Minardi da Cruz, J.; Singh, D.; Lamara, A.; Chebloune, Y. Small ruminant lentiviruses (SRLVs) break the species barrier to acquire new host range. Viruses 2013, 5, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Luciw, P. Human Immunodeficiency Viruses and Their Replication, 3rd ed.; Raven Press: Philadelphia, PA, USA, 1996; pp. 1881–1952. [Google Scholar]
- Vaishnav, Y.; Wong-Staal, F. The Biochemistry of AIDS. Annu. Rev. Biochem. 1991, 60, 577–630. [Google Scholar] [CrossRef] [PubMed]
- Kalland, K.H.; Szilvay, A.M.; Brokstad, K.A.; Saetrevik, W.; Haukenes, G. The human immunodeficiency virus type 1 Rev protein shuttles between the cytoplasm and nuclear compartments. Mol. Biol. Cell. 1994, 14, 7436–7444. [Google Scholar]
- Meyer, B.E.; Malim, M.H. The HIV-1 Rev trans-activator shuttles between the nucleus and the cytoplasm. Genes Dev. 1994, 8, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Daly, T.J.; Cook, K.S.; Gray, G.S.; Maione, T.E.; Rusche, J.R. Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature 1989, 342, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311. [Google Scholar] [PubMed]
- Simon, J.H.; Malim, M.H. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 1996, 70, 5297–5305. [Google Scholar] [PubMed]
- Fouchier, R.A.; Simon, J.H.; Jaffe, A.B.; Malim, M.H. Human immunodeficiency virus type 1 Vif does not influence expression or virion incorporation of gag-, pol-, and env-encoded proteins. J. Virol. 1996, 70, 8263–8269. [Google Scholar] [PubMed]
- Simon, J.H.; Miller, D.L.; Fouchier, R.A.; Malim, M.H. Virion incorporation of human immunodeficiency virus type-1 Vif is determined by intracellular expression level and may not be necessary for function. Virology 1998, 248, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Goila-Gaur, R.; Strebel, K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 2008. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: Not simply editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ikeuchi, K.; Byrn, R.; Groopman, J.; Baltimore, D. Lack of a negative influence on viral growth by the nef gene of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 9544–9548. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell. Biol. 2000, 2, 163–167. [Google Scholar] [PubMed]
- Atkins, K.M.; Thomas, L.; Youker, R.T.; Harriff, M.J.; Pissani, F.; You, H.; Thomas, G. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 2008, 283, 11772–11784. [Google Scholar] [CrossRef] [PubMed]
- Dikeakos, J.D.; Atkins, K.M.; Thomas, L.; Emert-Sedlak, L.; Byeon, I.J.; Jung, J.; Ahn, J.; Wortman, M.D.; Kukull, B.; Saito, M.; et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell. 2010, 21, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Thomas, L.; Ruby, C.E.; Atkins, K.M.; Morris, N.P.; Knight, Z.A.; Scholz, I.; Barklis, E.; Weinberg, A.D.; Shokat, K.M.; et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 2007, 1, 121–133. [Google Scholar] [CrossRef]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog 2008. [Google Scholar] [CrossRef] [PubMed]
- Swann, S.A.; Williams, M.; Story, C.M.; Bobbitt, K.R.; Fleis, R.; Collins, K.L. HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology 2001, 282, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Matthias, G.; Oliver, T.F.; Matija, P. Structure- function relationships in HIV-1 Nef. EMBO Rep. 2001, 2, 580–585. [Google Scholar]
- Roth, W.W.; Khan, M.; Geleziunas, R.; Stringer, H.G.; Zuberi, J.A.; Greene, W.C.; Powell, M.; Bond, V.C. Functionally-impaired HIV-1 Nef alleles from a mother-child transmission pair. Int. J. Mol. Sci. 2002, 3, 1058–1072. [Google Scholar] [CrossRef]
- Jere, A.; Fujita, M.; Adachi, A.; Nomaguchi, M. Role of HIV-1 Nef protein for virus replication in vitro. Microb. Infect. 2010, 12, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.; Lu, X.; Frost, J.; Geyer, M.; Jiang, B.; Luo, W.; Abo, A.; Alberts, A.; Peterlin, M. p21-activated kinase 1 plays a critical role in cellular activation by Nef. Mol. Biol. Cell. 2000, 20, 2619–2627. [Google Scholar] [CrossRef]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d'Aloja, P.; Schurmann, A.; Baur, S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Groschel, B.; Bushman, F. Cell cycle arrest in G2/M promotes early steps of infection by human immunodeficiency virus. J. Virol. 2005, 79, 5695–5704. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.A. From arrest to escape: HIV-1 Vpr cuts a deal. Cell Host Microbe 2014, 15, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Aida, Y. Visualizing Vpr-induced G2 arrest and apoptosis. PloS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Philippon, V.; Matsuda, Z.; Essex, M. Transactivation is a conserved function among primate lentivirus Vpr proteins but is not shared by Vpx. J. Hum. Virol. 1999, 2, 167–174. [Google Scholar] [PubMed]
- Mirani, M.; Elenkov, I.; Volpi, S.; Hiroi, N.; Chrousos, G.; Kino, T. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: Potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J. Immunol. 2002, 169, 6361–6368. [Google Scholar] [CrossRef] [PubMed]
- Hoch, J.; Lang, S.M.; Weeger, M.; Stahl-Hennig, C.; Coulibaly, C.; Dittmer, U.; Hunsmann, G.; Fuchs, D.; Muller, J.; Sopper, S.; et al. vpr deletion mutant of simian immunodeficiency virus induces AIDS in rhesus monkeys. J. Virol. 1995, 69, 4807–4813. [Google Scholar] [PubMed]
- Lang, S.M.; Weeger, M.; Stahl-Hennig, C.; Coulibaly, C.; Hunsmann, G.; Muller, J.; Muller-Hermelink, H.; Fuchs, D.; Wachter, H.; Daniel, M.M.; et al. Importance of vpr for infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 1993, 67, 902–912. [Google Scholar] [PubMed]
- Bouzar, A.B.; Guiguen, F.; Morin, T.; Villet, S.; Fornazero, C.; Garnier, C.; Gallay, K.; Gounel, F.; Favier, C.; Durand, J.; et al. Specific G2 arrest of caprine cells infected with a caprine arthritis encephalitis virus expressing vpr and vpx genes from simian immunodeficiency virus. Virology 2003, 309, 41–52. [Google Scholar]
- Bouzar, A.B.; Villet, S.; Morin, T.; Rea, A.; Genestier, L.; Guiguen, F.; Garnier, C.; Mornex, J.F.; Narayan, O.; Chebloune, Y. Simian immunodeficiency virus Vpr/Vpx proteins kill bystander noninfected CD4+ T-lymphocytes by induction of apoptosis. Virology 2004, 326, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Arrode-Brusés, G.; Halloway, N.; Narayan, O.; Chebloune, Y. Changes of biological properties and pathogenesis of CAEV chimeras expressing Nef and Vpx/Vpr accessory proteins in infected goats. Retrovirology 2009. [Google Scholar] [CrossRef]
- Cullen, B.R. HIV-1 auxiliary proteins: Making connections ina dying cell. Cell. 1998, 93, 685–692. [Google Scholar] [CrossRef]
- Emerman, M.; Malim, M. HIV-1 regulatory/accessory genes: Key to unraveling viral and host cell biology. Science 1998, 280, 1880–1884. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Harrich, D.; Pearson, L.; Mitsuyasu, R.; Gaynor, R. Functional domains required for Tat-induced transcriptional activation of the HIV-1 long terminal repeats. EMBO J. 1988, 7, 3143–3147. [Google Scholar] [PubMed]
- Hauber, J.; Malim, M.; Cullen, B.R. Mutational analysis of the conserved basic domain of human immunodeficiency virus Tat protein. J. Virol. 1989, 63, 1181–1187. [Google Scholar] [PubMed]
- Tang, H.; Kuhen, K.; Wong-Staal, F. Lentivirus replication and regulation. Annu. Rev. Genet. 1999, 33, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Guo, C.; Josephs, S.; Wong-Staal, F. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 1985, 229, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Saltarelli, M.J.; Schoborg, R.; Gdovin, S.L.; Clements, J.E. The CAEV tat gene transactivates the viral LTR and is necessary for efficient viral replication. Virology 1993, 197, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Neuveut, C.; Vigne, R.; Clements, J.E.; Sire, J. The visna transcriptional activator Tat: Effects on the viral LTR and on cellular genes. Virology 1993, 197, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Carruth, L.M.; Hardwick, J.; Morse, B.A.; Clements, J.E. Visna virus Tat protein: a potent transcription factor with both activator and suppressor domains. J. Virol. 1994, 68, 6137–6146. [Google Scholar] [PubMed]
- Villet, S.; Faure, C.; Bouzar, B.A.; Morin, T.; Verdier, G.; Chebloune, Y.; Legras, C. Lack of trans-activation function for Maedi Visna virus and Caprine arthritis encephalitis virus Tat proteins. Virology 2003, 307, 317–327. [Google Scholar] [CrossRef]
- Villet, S.; Bouzar, B.A.; Morin, T.; Verdier, G.; Legras, C.; Chebloune, Y. Maedi-Visna virus and caprine arthritis encephalitis virus genomes encode a Vpr-Like but No Tat protein. J. Virol. 2003, 77, 9632–9638. [Google Scholar] [CrossRef] [PubMed]
- Rea-Boutrois, A.; Villet, S.; Greenland, T.; Mehlen, P.; Chebloune, Y.; Verdier, G.; Legras-Lachuer, C. Small ruminant lentivirus Tat protein induces apoptosis in caprine cells in vitro by the intrinsic pathway. Virology 2009, 383, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rea-Boutrois, A.; Pontini, G.; Greenland, T.; Mehlen, P.; Chebloune, Y.; Verdier, G.; Legras-Lachuer, C. Caprine arthritis-encephalitis virus induces apoptosis in infected cells in vitro through the intrinsic pathway. Virology 2008, 375, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Delaugerre, C.; De Oliveira, F.; Lascoux-Combe, C.; Plantier, J.C.; Simon, F. HIV-1 group N: Travelling beyond Cameroon. Lancet 2011, 378, 1894. [Google Scholar] [CrossRef]
- Plantier, J.C.; Leoz, M.; Dickerson, J.E.; De Oliveira, F.; Cordonnier, F.; Lemee, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Vallari, A.; Holzmayer, V.; Harris, B.; Yamaguchi, J.; Ngansop, C.; Makamche, F.; Mbanya, D.; Kaptue, L.; Ndembi, N.; Gurtler, L.; et al. Confirmation of putative HIV-1 group P in Cameroon. J. Virol. 2011, 85, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- D’Arc, M.; Ayouba, A.; Esteban, A.; Learn, G.H.; Boue, V.; Liegeois, F.; Etienne, L.; Tagg, N.; Leendertz, F.H.; Boesch, C.; et al. Origin of the HIV-1 group O epidemic in western lowland gorillas. Proc. Natl. Acad. Sci. USA 2015, 112, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Touloumi, G.; Hatzakis, A. Natural history of HIV-1 infection. Clin. Dermatol. 2000, 18, 389–399. [Google Scholar] [CrossRef]
- Mitchell, D.T. Investigations into Jaagziekte or Chronic Catarrhal Pneumonia of Sheep; Directory Veterinary Education Research: Union of South Africa, UK, 1915; p. 585. [Google Scholar]
- Palsson, P.A. Maedi and visna in sheep. Front. Biol. 1976, 44, 17–43. [Google Scholar] [PubMed]
- Marsh, H. Progressive pneumonia in sheep. J. Am. Vet. Med. Assoc. 1923, 62, 458–473. [Google Scholar]
- Gislason, G. Thaettir urn Indutning Bdfjar og Karak~Ilsjdkdbma; Icelandic Department of Agricultural Publication: Reykjavik, Iceland, 1947; pp. 235–254. (In Icelandic)
- Palsson, P.A. Maedi-visna. J. Clin. Pathol. 1972, 6, 115–120. [Google Scholar] [CrossRef]
- Stunzi, H.; Buchi, H.F.; LeRoy, H.L.; Leeman, W. Endemische arthritis chronic a bei zeiegen. Schw. Arch. Tier. 1959, 106, 778–788. [Google Scholar]
- Stavrou, D.; Deutschlander, N.; Dahme, E. Granulomatous encephalomyelitis in goats. J. Comp. Pathol. 1969, 79, 393–396. [Google Scholar] [CrossRef]
- Cork, L.C.; Hadlow, W.J.; Crawford, T.B.; Gorham, J.R.; Piper, R.C. Infectious leukoencephalomyelitis of young goats. J. Infect. Dis. 1974, 129, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.B.; Adams, D.S.; cheevers, W.; Cork, L.C. Chronic arthritis in goats caused by a retrovirus. Science 1980, 207, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Narayan, O.; Clements, J.E.; Strandberg, J.D.; Cork, L.C.; Griffin, D.E. Biological characterization of the virus causing leukoencephalitis and arthritis in goats. J. Gen. Virol. 1980, 50, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Peluso, R.; Haase, A.; Stowring, L.; Edwards, M.; ventura, P. A trojan hosre mechanism for the spread of visna virus in monocytes. Virology 1985, 147, 231–236. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Narayan, O.; Molineaux, S.; Clements, J.E.; Ghotbi, Z. Slow, persistent replication of lentiviruses: role of tissue macrophages and macrophage precursors in bone marrow. Proc. Natl. Acad. Sci. USA 1985, 82, 7086–7090. [Google Scholar] [CrossRef] [PubMed]
- Lairmore, M.D.; Akita, G.Y.; Russell, H.I.; DeMartini, J.C. Replication and cytopathic effects of ovine lentivirus strains in alveolar macrophages correlate with in vivo pathogenicity. J. Virol. 1987, 61, 4038–4042. [Google Scholar] [PubMed]
- Narayan, O.; Kennedy-Stoskopf, S.; Sheffer, D.; Griffin, D.E.; Clements, J.E. Activation of caprine arthritis-encephalitis virus expression during maturation of monocytes to macrophages. Infect. Immun. 1983, 41, 67–73. [Google Scholar] [PubMed]
- Blacklaws, B.A. Small ruminant lentiviruses: immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.L.; Mazzei, M.; Bandecchi, P.; Arispici, M.; Tolari, F. In situ PCR-associated immunohistochemistry identifies cell types harbouring the Maedi-Visna virus genome in tissue sections of sheep infected naturally. J. Virol. Methods 2003, 107, 121–127. [Google Scholar] [CrossRef]
- Sanna, E.; Sanna, M.P.; Vitali, C.G.; Renzoni, G.; Sanna, L.; Spano, S.; Rossi, G.; Leoni, A. Proviral DNA in the brains of goats infected with caprine arthritis-encephalitis virus. J. Comp. Pathol. 1999, 121, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Pautrat, G.; Filippi, P. Evidence for the production of a fusion factor during in vitro infection of sheep choroid plexus cells by visna virus. C. R. Seances Soc. Biol. Fil. 1979, 173, 811–817. [Google Scholar] [PubMed]
- Sihvonen, L.; Veijalainen, P. Kinetics of maedi virus production in sheep choroid plexus cells. Vet. Microbiol. 1981, 6, 1–8. [Google Scholar] [CrossRef]
- Ryan, S.; Tiley, L.; McConnell, I.; Blacklaws, B. Infection of dendritic cells by the Maedi-Visna lentivirus. J. Virol. 2000, 74, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Lechat, E.; Milhau, N.; Brun, P.; Bellaton, C.; Greenland, T.; Mornex, J.F.; le Jan, C. Goat endothelial cells may be infected in vitro by transmigration of caprine arthritis-encephalitis virus-infected leucocytes. Vet. Immunol. Immunopathol. 2005, 104, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, H.; Koot, M.; Kootstra, N.A.; Dercksen, M.W.; de Goede, R.E.; van Steenwijk, R.P.; Lange, J.M.; Schattenkerk, J.K.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar] [PubMed]
- McCune, J.M. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 2001, 410, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Abbas, W.; Varin, A.; Kumar, A.; Di Martino, V.; Dichamp, I.; Herbein, G. HIV-1 Nef interacts with HCV Core, recruits TRAF2, TRAF5 and TRAF6, and stimulates HIV-1 replication in macrophages. J. Innate Immun 2013, 5, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Zhang, H.; Lopez, P.; Pardo, C.A.; Gartner, S. In vitro modeling of the HIV-1 macrophage reservoir. J. Leukoc. Biol. 2006, 80, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; Calio, R.; Balzarini, J.; Bellocchi, M.; Garaci, E.; Perno, C. Macrophages and HIV infection: therapeutical approaches toward this strategic virus reservoir. Antiviral Res. 2002, 55, 209–225. [Google Scholar] [CrossRef]
- Crowe, S.; Zhu, T.; muller, W. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J. Leukoc. Biol. 2003, 74, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Lambotte, O.; Taoufik, Y.; de Goer, M.; Wallon, C.; Goujard, C.; Delfraissy, J. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2000, 23, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for Human Immunodeficiency Virus Type 1 Replication In Vivo in CD14+ Monocytes and Its Potential Role as a Source of Virus in Patients on Highly Active Antiretroviral Therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010. [Google Scholar] [CrossRef] [PubMed]
- Zink, M.C.; Yager, J.A.; Myers, J.D. Pathogenesis of caprine arthritis encephalitis virus. Cellular localization of viral transcripts in tissues of infected goats. Am. J. Pathol. 1990, 136, 843. [Google Scholar] [PubMed]
- Clements, J.E.; Zink, M.C.; Narayan, O.; Gabuzda, D. Lentiviral infection of macrophages. Immunol. Ser. 1994, 60, 589–600. [Google Scholar]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009. [Google Scholar] [CrossRef] [PubMed]
- Lore, K.; Smed-sorensen, A.; Vasudevan, J.; Mascola, J.; Koup, R.A. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J. Exp. Med. 2005, 201, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Smed-Sörensen, A.; Loré, K.; Vasudevan, J.; Louder, M.K.; Andersson, J.; Mascola, J.R.; Spetz, A.-L.; Koup, R.A. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol. 2005, 79, 8861–8869. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, L.; Lozach, P.-Y.; Schiffer, C.; Staropoli, I.; Pezo, V.; Porrot, F.; Canque, B.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Amara, A. Infection of dendritic cells (DCs), not DC-SIGN-mediated internalization of human immunodeficiency virus, is required for long-term transfer of virus to T cells. J. Virol. 2006, 80, 2949–2957. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Evans, V.A.; Kumar, N.; Filali, A.; Procopio, F.A.; Yegorov, O.; Goulet, J.P.; Saleh, S.; Haddad, E.K.; da Fonseca Pereira, C.; Ellenberg, P.C.; et al. Myeloid dendritic cells induce HIV-1 latency in non-proliferating CD4+ T cells. PLoS Pathog. 2013, 9, e1003799. [Google Scholar] [CrossRef] [PubMed]
- Banks, K.L.; Adams, D.S.; McGuire, T.C.; Carlson, J. Experimental infection of sheep by caprine arthritis-encephalitis virus and goats by progressive pneumonia virus. Am. J. Vet. Res. 1983, 44, 2307–2311. [Google Scholar] [PubMed]
- Leroux, C.; Cruz, J.C.; Mornex, J.F. SRLVs: A genetic continuum of lentiviral species in sheep and goats with cumulative evidence of cross species transmission. Curr. HIV Res. 2010, 8, 94–100. [Google Scholar] [PubMed]
- Karr, B.M.; Chebloune, Y.; Leung, K.; Narayan, O. Genetic characterization of two phenotypically distinct North American ovine lentiviruses and their possible origin from caprine arthritis-encephalitis virus. Virology 1996, 225, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.; Huder, J.B.; Boni, J.; Schonmann, M.; Muhlherr, J.; Lutz, H.; Schupbach, J. Direct evidence for natural transmission of small-ruminant lentiviruses of subtype A4 from goats to sheep and vice versa. J. Virol. 2004, 78, 7518–7522. [Google Scholar] [CrossRef] [PubMed]
- Guiguen, F.; Mselli-Lakhal, L.; Durand, J.; Du, J.; Favier, C.; Fornazero, C.; Grezel, D.; Balleydier, S.; Hausmann, E.; Chebloune, Y. Experimental infection of Mouflon-domestic sheep hybrids with caprine arthritis-encephalitis virus. Am. J. Vet. Res. 2000, 61, 456–461. [Google Scholar] [CrossRef]
- Morin, T.; Guiguen, F.; Bouzar, B.A.; Villet, S.; Greenland, T.; Grezel, D.; Gounel, F.; Gallay, K.; Garnier, C.; Durand, J.; et al. Clearance of a Productive Lentivirus Infection in Calves Experimentally Inoculated with Caprine Arthritis-Encephalitis Virus. J. Virol. 2003, 77, 6430–6437. [Google Scholar] [CrossRef] [PubMed]
- Erhouma, E.; Guiguen, F.; Chebloune, Y.; Gauthier, D.; Lakhal, L.M.; Greenland, T.; Mornex, J.F.; Leroux, C.; Alogninouwa, T. Small ruminant lentivirus proviral sequences from wild ibexes in contact with domestic goats. J. Gen. Virol. 2008, 89, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, G.; Bertoni, G.; Puricelli, M.; Maccalli, M.; Moroni, P. Demonstration of coinfection with and recombination by caprine arthritis-encephalitis virus and maedi-visna virus in naturally infected goats. J. Virol. 2007, 81, 4948–4955. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.S.; Klevjer-Anderson, P.; Carlson, J.L.; McGuire, T.C.; Gorham, J.R. Transmission and control of caprine arthritis-encephalitis virus. Am. J. Vet. Res. 1983, 44, 1670–1675. [Google Scholar] [PubMed]
- Knight, A.; Jokinen, M. Caprine arthritis-encephalitis. Compend. Contin. Educ. Pract. Vet. 1982, 4, 263–269. [Google Scholar]
- Lloyd, S. Goat medicine and surgery. Br. Vet. J. 1982, 138, 70–85. [Google Scholar] [PubMed]
- Sherman, D.M. CAE: Caprine arthritis encephalitis—a growing concern. DGJ 1983, 61, 93–95. [Google Scholar]
- Preziuso, S.; Renzoni, G.; Allen, T.E.; Taccini, E.; Rossi, G.; DeMartini, J.C.; Braca, G. Colostral transmission of maedi visna virus: sites of viral entry in lambs born from experimentally infected ewes. Vet. Microbiol. 2004, 104, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Cork, L.C.; Narayan, O.; Strandberg, J.; Clements, J.E.; Griffin, D. Viral leukoencephalomyelitis-arthritis of goats: Pathogenesis of the persistent viral infection. J. Neuropath. Exp. Neur. 1980. [Google Scholar] [CrossRef]
- Rimstad, E.; East, N.E.; Torten, M.; Higgins, J.; DeRock, E.; Pedersen, N.C. Delayed seroconversion following naturally acquired caprine arthritis-encephalitis virus infection in goats. Am. J. Vet. Res. 1993, 54, 1858–1862. [Google Scholar] [PubMed]
- Cork, L.C. Pathology and epidemiology of lentiviral infection of goats. In Maedi-Visna and Related Diseases; Pétursson, G., Hoff-Jørgensen, R., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1990; pp. 119–127. [Google Scholar]
- Daas, E.; Moudgil, T.; Meyer, R. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N. Engl. J. Med. 1991, 324, 961–964. [Google Scholar]
- Piatak, M.; saag, M.; yang, L. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 1993, 259, 1749–1754. [Google Scholar] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Saksena, N.; Potter, S. Reservoirs of HIV-1 in vivo: implications for antiretroviral therapy. AIDS Rev. 2003, 5, 3–18. [Google Scholar] [PubMed]
- Busch, M.; El-Amad, Z.; Sheppard, H.; Ascher, M. Primary HIV-1 infection. N. Engl. J. Med. 1991, 325, 733–735. [Google Scholar]
- Clark, S.; Saag, M.; Decker, W. High titers of cytopathic virus in plasma of patients with symptomatic primary infection. N. Engl. J. Med. 1991, 324, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Tindall, B.; Cooper, D. Primary HIV infection: Host responses and intervention strategies. AIDS 1991, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kingsley, L.; Armstrong, J.; Ding, M.; Cottrill, M.; Rinaldo, C. Enhanced expression of human immunodeficiency virus type 1 correlates with development of AIDS. Virology 1993, 196, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Henrad, D.; Philips, J.; Muenz, L.; Blattner, W.A.; Wiesner, D.; Eyster, M.E. Natural history of HIV-1 cell free viremia. J. Am. Med. Assoc. 1995, 274, 554–558. [Google Scholar] [CrossRef]
- Jurriaans, S.; van German, B.; Weverling, G.J.; van Strijp, D.; Nara, P.; Coutinho, R.; Koot, M.; Schuitemaker, H.; Goudsmit, J. The natural history of HIV-1 infection: Virus load and virus phenotype independent determinants of clinical course? Virology 1994, 204, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Cozzi-Lepri, A.; Sabin, C.; Tezzotti, P. Is there a general tendency for rate of CD41 lymphocyte count decline to speed up during HIV infection? Evidence from the Italian Seroconversion Study. J. Infect. Dis. 1997, 175, 775–780. [Google Scholar] [CrossRef]
- Lang, W.; Perkins, H.; Anderson, R.; Royce, R.; Jewell, N. Patterns of T lymphocyte changes with human immunodeficiency virus infection: From seroconversion to the development of AIDS. J. Acquir. Immune Defic. Syndr. 1989, 2, 63–69. [Google Scholar] [PubMed]
- Phillips, A.N.; Lee, C.A.; Elford, J.; Janossy, G.; Timms, A.; Bofill, M.; Kernoff, P.B. Serial CD4 lymphocyte counts and development of AIDS. Lancet 1991, 337, 389–392. [Google Scholar] [PubMed]
- Touloumi, G.; Hatzakis, A.; Rosenberg, P.S.; O’Brien, T.R.; Goedert, J.J. Effects of age at seroconversion and baseline HIV-RNA level on the loss of CD41 cells among persons with hemophilia. AIDS 1998, 12, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Lewthwaite, P.; Wilkins, E. Natural history of HIV/AIDS. Medicine 2005, 33, 10–13. [Google Scholar] [CrossRef]
- Lewthwaite, P.; Wilkins, E. Natural history of HIV/AIDS. Medicine 2009, 37, 333–337. [Google Scholar] [CrossRef]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, San Francisco city cohort, ALIVE study. Science 1996, 273, 1856–1862. [Google Scholar] [PubMed]
- Al-Jabri, A.A. Mechanisms of host resistance against HIV infection and progression to AIDS. Sultan Qaboos Univ. Med. J. 2007, 7, 82–96. [Google Scholar] [PubMed]
- Crowe, S.; Carlin, J.; Steward, K.; Lucas, C.R. Predictive value of CD4 lymphocyte numbers for the development of opportunistic infections and malignacies in HIV-infected persons. J. Acquir. Immune Defic. Syndr. 1991, 4, 770–776. [Google Scholar] [PubMed]
- Hoover, D.R.; Rinaldo, C.; He, Y.; Phair, J.; Fahey, J.; Graham, N.M. Long-term survival without clinical AIDS after CD4+ cell fall below 200 × 106/L. AIDS 1995, 9, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Mocroft, A.; Johnson, M.; Sabin, C.; Lipman, M.; Elford, J.; Emery, V.; Morcinek, J.; Youle, M.; Janossy, G.; Lee, C.A. Staging system for clinical AIDS patients. Lancet 1995, 346, 12–17. [Google Scholar] [CrossRef]
- Orenstein, R. Presenting Syndromes of Human Immunodeficiency Virus; Elsevier: Amsterdam, The Netherlands, 2002; pp. 1097–1102. [Google Scholar]
- O’Sullivan, B.M.; Eaves, F.W.; Baxendell, S.A.; Rowan, K.J. Leucoencephalomyelitis of goat kids. Aust. Vet. J. 1978, 54, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.S.; Crawford, T.B.; Klevjer-Anderson, P. A pathogenetic study of the early connective tissue lesions of viral caprine arthritis-encephalitis. Amer. J. Pathol. 1980, 99, 257–278. [Google Scholar]
- Sigurdsson, B.; Palsson, P.A. Visna of sheep. A slow demyelinating infection. Brit. J. Exp. Pathol. 1958, 39, 519–528. [Google Scholar]
- Ruprecht, R.M.; Baba, T.W.; Liska, V.; Ray, N.B.; Martin, L.N.; Murphey-Corb, M.; Rizvi, T.A.; Bernacky, B.J.; Bernacky, M.E.; McClure, H.M.; et al. Oral transmission of primate lentiviruses. J. Infect. Dis. 1999, 179, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A.; et al. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.; Betjes, M.; Romani, N.; Hirmand, H.; Cameron, P.; Hoffman, L.; Gezelter, S.; Schuler, G.; Steinman, R. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell. 1994, 78, 389–398. [Google Scholar] [CrossRef]
- Neutra, M.; Frey, A.; Kraehenbuhl, J. Epithelial M cells: Gateways for mucosal infection and immunization. Cell. 1996, 86, 345–348. [Google Scholar] [CrossRef]
- Hladik, F.; Hope, T.J. HIV infection of the genital mucosa in women. Curr. HIV/AIDS Rep. 2009, 6, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Stieh, D.J.; Maric, D.; Kelley, Z.L.; Anderson, M.R.; Hattaway, H.Z.; Beilfuss, B.A.; Rothwangl, K.B.; Veazey, R.S.; Hope, T.J. Vaginal challenge with an SIV-based dual reporter system reveals that infection can occur throughout the upper and lower female reproductive tract. PLoS Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, S. Pathogenesis of HIV in the Gastrointestinal tract. Curr HIV/AIDS Rep. 2007, 4, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S. Getting to the Guts of HIV Pathogenesis. J. Exp. Med. 2004, 200, 697–700. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.; Spencer, J. Lymphoid cells and tissues of the gastrointestinal tract; Cambridge University Press: Cambridge, UK, 1994; pp. 1–23. [Google Scholar]
- Schieferdecker, H.; Ullrich, R.; Hirseland, H.; Zeitz, M. T cell differentiation antigens on lymphocytes in the human intestinal lamina propria. J. Immunol. 1992, 149, 2816–2822. [Google Scholar] [PubMed]
- Brenchley, J.; Schacker, T.; Ruff, L.; Price, D.A.; Taylor, J.H.; Beilman, G.J.; Nguyen, P.L.; Khoruts, A.; Larson, M.; Haase, A.T.; et al. CD4+T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 2004, 200, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S.; Marx, P.; Lackner, A. Importance of the state of activation and/or differentiation of CD4+ T cells in AIDS pathogenesis. Trends Immunol. 2002, 23, 128–129. [Google Scholar] [CrossRef]
- Mattapallil, J.; Douek, D.; Hill, B.; Nishimura, Y.; Martin, M.; Roederer, M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 2005, 434, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Lackner, A.A.; Mohan, M.; Veazey, R.S. The Gastrointestinal Tract and AIDS Pathogenesis. Gastroenterology 2009, 136, 1966–1978. [Google Scholar] [CrossRef]
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV pathogenesis: The host. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Katsikis, P.D.; Mueller, Y.M.; Villinger, F. The cytokine network of acute HIV Infection: A promising target for vaccines and therapy to reduce viral set-point? PLoS Pathog. 2011. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV Infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Elbirt, D.; Mahlab-Guri, K.; Bezalel-Rosenberg, S.; Gill, H.; Attali, M.; Asher, I. HIV-associated neurocognitive disorders (HAND). Isr. Med. Assoc. J. 2015, 17, 54–59. [Google Scholar]
- Albright, A.V.; Soldan, S.S.; Gonzalez-Scarano, F. Pathogenesis of human immunodeficiency virus-induced neurological disease. J. Neurovirol. 2003, 9, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Lipton, S.A. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 8212–8216. [Google Scholar] [CrossRef] [PubMed]
- Lindl, K.A.; Marks, D.R.; Kolson, D.L.; Jordan-Sciutto, K.L. HIV-associated neurocognitive disorder: Pathogenesis and therapeutic opportunities. J. Neuroimmune Pharmacol. 2010, 5, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Price, R.W.; Brew, B.; Sidtis, J.; Rosenblum, M.; Scheck, A.C.; Cleary, P. The brain in AIDS: Central nervous system HIV-1 infection and AIDS dementia complex. Science 1988, 239, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Valcour, V.; Sithinamsuwan, P.; Letendre, S.; Ances, B. Pathogenesis of HIV in the Central Nervous System. Curr. HIV/AIDS Rep. 2011, 8, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Fieni, F.; Rowe, J.; Van Hoosear, K.; Burucoa, C.; Oppenheim, S.; Anderson, G.; Murray, J.; BonDurant, R. Presence of caprine arthritis-encephalitis virus (CAEV) proviral DNA in genital tract tissues of superovulated dairy goat does. Theriogenology 2003, 59, 1515–1523. [Google Scholar] [CrossRef]
- Petursson, G.; Nathanson, N.; Georgsson, G.; Panitch, H.; Palsson, P.A. Pathogenesis of visna Sequential virologic, serologic and pathologic studies. Lab. Invest. 1976, 35, 402–412. [Google Scholar] [PubMed]
- Adams, D.S.; Crawford, T.B.; Klevjer-Anderson, P. A pathogenetic study of the early connective tissue lesions of viral caprine arthritis-encephalitis. Am. J. Pathol. 1980, 99, 257–278. [Google Scholar]
- Narayan, O.; Cork, L.C. Lentiviral diseases of sheep and goats: Chronic pneumonia leukoencephalomyelitis and arthritis. Rev. Infect. Dis. 1985, 7, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Cork, L.C.; Davis, W.C. Ultrastructural features of viral leukoencephalomyelitis of goats. Lab. Invest. 1975, 32, 359–365. [Google Scholar] [PubMed]
- Ellis, T.M.; Robinson, W.F.; Wilcox, G.E. The pathology and aetiology of lung lesions in goats infected with caprine arthritis-encephalitis virus. Aust. Vet. J. 1988, 65, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, B.; Grimsson, H.; Palsson, P.A. Maedi, a chronic, progressive infection of sheep’s lungs. J. Infect. Dis. 1952, 90, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Cutlip, R.C.; Lehmkuhl, H.D.; Brogden, K.A.; Bolin, S.R. Mastitis associated with ovine progressive pneumonia virus infection in sheep. Am. J. Vet. Res. 1985, 46, 326–328. [Google Scholar] [PubMed]
- Clapham, P.R.; McKnight, A. HIV-1 receptors and cell tropism. Brit. Med. Bull. 2001, 58, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Michael, N. Host genetic influences on HIV-1 pathogenesis. Curr. Opin. Immunol. 1999, 11, 466–474. [Google Scholar] [CrossRef]
- Crespo, H.; Jáuregui, P.; Glaria, I.; Sanjose, L.; Polledo, L.; García-Marin, J.; Luján, L.; de Andrés, D.; Amorena, B.; Reina, R. Mannose receptor may be involved in small ruminant lentivirus pathogenesis. Vet. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Reina, R.; Glaria, I.; Ramírez, H.; de Andrés, X.; Jáuregui, P.; Luján, L.; Martinez- Pomares, L.; Amorena, B.; de Andrés, D. Identification of the ovine mannose receptor and its possible role in Visna/Maedi virus infection. Vet. Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Narayan, O.; Wolinsky, J.; Clements, J.E.; Strandberg, J.; Griffin, D.; Cork, L.C. Slow virus replication: The role of macrophages in the persistence and expression of visna viruses of sheep and goats. J. Gen. Virol. 1982, 59, 345–356. [Google Scholar] [CrossRef]
- Chen, M.; Westmoreland, S.; Ryzhova, E.; MArtin, G.J.; Soldan, S.; Lackner, A.A.; González-Scarano, F. Simian immunodeficiency virus envelope compartmentalizes in brain regions independent of neuropathology. J. Neurovirol. 2006, 12, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Hudson, L.; Tompkins, M.; Vahlenkamp, T.; Meeker, R. Compartmentalization and evolution of feline immunodeficiency virus between the central nervous system and periphery following intracerebroventricular or systemic inoculation. J. Neurovirol. 2006, 12, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Kosakovsky, P.; Shapshak, F. Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. J. Virol. 2007, 81, 6643–6651. [Google Scholar] [CrossRef]
- Ramirez, H.; Reina, R.; Bertolotti, L.; Cenoz, A.; Hernandez, M.; San Roman, B.; Glaria, I.; de Andrés, X.; Crespo, H.; Jauregui, P. Study of compartmentalization in the visna clinical form of small ruminant lentivirus infection in sheep. BMC Vet. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Kunstman, K.; Patterson, B.; Furtado, M.; McEvilly, M.; Levy, R.; Wolinsky, S. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type-1 infected patients: Evidence of conserved elements in the V3 region of the Envelope protein of brain-derived sequences. J. Virol. 1994, 68, 7467–7481. [Google Scholar]
- Hotzel, I.; Cheevers, W. Sequence similarity between the envelope surface unit (SU) glycoproteins of primate and small ruminant lentiviruses. Virus Res. 2000, 69, 47–54. [Google Scholar] [CrossRef]
- Mwaengo, D.; Grant, R.; DeMartini, J.; Carlson, J. Envelope glycoprotein nucleotide sequence and genetic characterization of North American ovine lentiviruses. Virology 1997, 238, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Blackard, J.T. HIV compartmentalization: a review on a clinically important phenomenon. Curr HIV Res. 2012, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; di Mascio, M.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Wrin, T.; Liegler, T.; Hoh, R.; Hayden, M.; Barbour, J.D.; Hellmann, N.S.; Petropoulos, C.J.; McCune, J.M.; Hellerstein, M.K.; et al. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. New Engl. J. Med. 2001, 344, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Rezk, N.L.; Ince, W.L.; Cheema, M.; Zhang, L.; Su, L.; Swanstrom, R.; Kashuba, A.D.; Margolis, D.M. Suppression of human immunodeficiency virus type 1 (HIV-1) viremia with reverse transcriptase and integrase inhibitors, CD4+ T-cell recovery, and viral rebound upon interruption of therapy in a new model for HIV treatment in the humanized Rag2-/-γc-/- mouse. J. Virol. 2009, 83, 8254–8258. [Google Scholar] [PubMed]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Hjelle, B.L.; Miller, V.E.; Palmer, D.L.; Llewellyn, A.L.; Merlin, T.L.; Young, S.A.; Mills, R.G.; Wachsman, W.; Wiley, C.A. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 1992, 42, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Fauci, A.S. Latent reservoirs of HIV: obstacles to the eradication of virus. Proc. Natl. Acad. Sci. USA 1999, 96, 10958–10961. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Fauci, A.S. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Samuel, W.; Warner, G. Host factor regulating post integration latency of HIV. Trends Microbiol. 2005, 13, 137–139. [Google Scholar]
- du Chéné, I.; Basyuk, E.; Lin, Y.-L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for therapeutic strategies. Retrovirology 2012. [Google Scholar] [CrossRef]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Siliciano, R.F. From reactivation of latent HIV-1 to elimination of the latent reservoir: The presence of multiple barriers to viral eradication. Bioessays 2013, 35, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the search for a cure for HIV-1 infection: Targeting the latent reservoir for HIV-1. J. Allergy Clin. Immun. 2014, 134, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.; Han, Y.; Zhou, Y.; Siliciano, J.; Siliciano, R.F. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 2004, 10, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sharp, P. Novel mechanisms and factors for regulation by HIV-1 Tat. EMBO J. 1995, 14, 321–328. [Google Scholar] [PubMed]
- Jordan, A.; Defechereux, P.; Verdin, E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Osorio, A.A.; Munoz, A.; Torres-Romero, D.; Bedoya, L.M.; Perestelo, N.R.; Jimenez, I.A.; Alcami, J.; Bazzocchi, I.L. Olean-18-ene triterpenoids from Celastraceae species inhibit HIV replication targeting NF-kB and Sp1 dependent transcription. Eur. J. Med. Chem. 2012, 52, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, D.H.; Ganesh, K.N.; Mitra, D. HIV-1 Tat directly binds to NFκB enhancer sequence: Role in viral and cellular gene expression. Nucleic Acids Res. 2004, 32, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Somasundaran, M.; Green, M.R. Human transcription factor YY1 represses human immunodeficiency virus type 1 transcription and virion production. J. Virol. 1994, 68, 905–910. [Google Scholar] [PubMed]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lin, Y.; An, W.; Xu, J.; yahg, H.; O’Connell, K.; Dordai, D.; Boeke, J.; Siliciano, J.D.; Siliciano, R.F. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.M.; Amaral, M.C.; Wu, J.Y.; Maniatis, T.; Greene, W.C. HIV Rev-dependent binding of SF2/ASF to the Rev response element: possible role in Rev-mediated inhibition of HIV RNA splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.; Kashanchi, F. Tat gets the “green” light on transcription initiation. Retrovirology 2005. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Peterlin, B.M. A New Paradigm in Eukaryotic Biology: HIV Tat and the Control of Transcriptional Elongation. PloS Biol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.; Stowring, L.; Narayan, O.; Griffin, D.; Price, D. Slow persistant infection caused by visna virus role of host restriction. Science 1977, 195, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Thormar, H. Physical, Chemical, Biological Properties of Visna Virus and Its Relationship to Other Animal Viruses. In Slow, Latent and Temprate Virus Infection; The National Institute of Neurological Disorders and Blindness: Washington, DC, USA, 1966; pp. 335–340. [Google Scholar]
- Thormar, H. Maedi-visna virus and its relationship to human immunodeficiency virus. AIDS Rev. 2005, 7, 233–245. [Google Scholar] [PubMed]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Gracia, J.; Harrich, D.; Soultanakis, E.; Wu, F.; Mitsuyasu, R.; Gaynor, R. Human immunodeficiency virus type 1 LTR TATA and TAR region sequences required for transcriptional regulation. EMBO J. 1989, 8, 765–778. [Google Scholar]
- Harrich, D.; Gracia, J.; Mitsuyasu, R.; Gaynor, R. TAR independent activation of the human immunodeficiency virus in phorbol ester stimulated T lymphocytes. EMBO J. 1990, 9, 4417–4423. [Google Scholar] [PubMed]
- Olsen, H.S.; Rosen, C. Contribution of the TATA motif to Tat-mediated transcriptional activation of the human immunodeficiency virus gene expression. J. Virol. 1992, 66, 5594–5597. [Google Scholar] [PubMed]
- Danino, Y.M.; Even, D.; Ideses, D.; Juven-Gershon, T. The core promoter: At the heart of gene expression. Biochim. Biophys. Acta 2015, 1849, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Ooms, M.; Malcolm, T.; Simon, V.; Sadowski, I. Identification and functional analysis of a second RBF-2 binding site within the HIV-1 promoter. Virology 2011, 418, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, E.; Doyle, M.C.; Nzaramba, I.; Magdzinski, A.; Dumais, N.; Bell, B. CTGC motifs within the HIV core promoter specify Tat-responsive pre-initiation complexes. Retrovirology 2012. [Google Scholar] [CrossRef] [PubMed]
- Miller-Jensen, K.; Skupsky, R.; Shah, P.S.; Arkin, A.P.; Schaffer, D.V. Genetic selection for context-dependent stochastic phenotypes: Sp1 and TATA mutations increase phenotypic noise in HIV-1 gene expression. PLoS Comput. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Duverger, A.; Jones, J.; May, J.; Bibollet-Ruche, F.; Wagner, F.A.; Cron, R.Q.; Kutsch, O. Determinants of the establishment of human immunodeficiency virus type 1 latency. J. Virol. 2009, 83, 3078–3093. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Rittner, K. The human immunodeficiency virus long terminal repeat includes a specialised initiator element which is required for Tat responsive transcription. J. Mol. Biol. 1995, 248, 562–580. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.K.; Buckler-White, A.J.; Rabson, A.B.; Englund, G.; Martin, M.A. Contribution of NF-κB and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: Distinct patterns of viral growth are determined by T-cell types. J. Virol. 1991, 65, 4350–4358. [Google Scholar] [PubMed]
- Dahmus, M. Phosphorylation of C-terminal domain of RNA polymerase II. Biochim. Biophys. Acta 1995, 171–182. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Biol. Cell. 2002, 22, 4622–4637. [Google Scholar] [CrossRef]
- Guo, J.; Price, D.H. RNA polymerase II transcription elongation control. Chem. Rev. 2013, 113, 8583–8603. [Google Scholar] [CrossRef]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Biol. Cell. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, M.; Schiralli Lester, G.M.; Lee, C.; Missra, A.; Wasserman, G.A.; Steffen, M.; Gilmour, D.S.; Henderson, A.J. Negative elongation factor (NELF) coordinates RNA polymerase II pausing, premature termination, and chromatin remodeling to regulate HIV transcription. J. Biol. Chem. 2013, 288, 25995–26003. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Price, D.H. Analysis of factor interactions with RNA polymerase II elongation complexes using a new electrophoretic mobility shift assay. Nucleic Acids Res. 2008, 36, e135. [Google Scholar] [CrossRef] [PubMed]
- Missra, A.; Gilmour, D.S. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11301–11306. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA binding in HIV-1 and promoter-proximal pause regions. PLoS Genet. 2014. [Google Scholar] [CrossRef]
- Feng, S.; Holland, E.C. HIV-1 Tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Babayeva, N.D.; Suwa, Y.; Baranovskiy, A.G.; Price, D.H.; Tahirov, T.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell. Cycle 2014, 13, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Rice, A. Lentivirus Tat proteins specifically associate with a cellular protein- kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J. Virol. 1995, 69, 1612–1620. [Google Scholar] [PubMed]
- Montanuy, I.; Torremocha, R.; Hernandez-Munain, C.; Sune, C. Promoter influences transcription elongation: TATA-box element mediates the assembly of processive transcription complexes responsive to cyclin-dependent kinase 9. J. Biol. Chem. 2008, 283, 7368–7378. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, T.; Price, D.H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef] [PubMed]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.F.; Price, D. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol. Biol. Cell. 1992, 12, 2078–2090. [Google Scholar]
- Fujinaga, K. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Biol. Cell. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Mbonye, U.R.; Gokulrangan, G.; Datt, M.; Dobrowolski, C.; Cooper, M.; Chance, M.R.; Karn, J. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6, 1715–1758. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. Tat, A Novel Regulator of HIV Transcription and Latency. In HIV Sequence Compendium 2000; Kuiken, C.M.F., Foley, B., Mellors, J.W., Hahn, B., Mullins, J., Marx, P., Wolinsky, S., Eds.; Theoretical Biology and Biophysics Group Los Alamos National Laboratory: Los Alamos, NM, USA, 2000; pp. 2–18. [Google Scholar]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Strebel, K. Virus-host interactions: role of HIV proteins Vif, Tat, and Rev. AIDS 2003, 17, 25–34. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, M. Lost in Transcription: Molecular Mechanisms that Control HIV Latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Schiralli Lester, G.M.; Henderson, A.J. Mechanisms of HIV transcriptional regulation and their contribution to latency. Mol. Biol. Int. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Gdovin, S.; Clements, J.E. Molecular mechanisms of visna virus Tat: Identification of the targets for transcriptional activation and evidence for a post-transcriptional effect. Virology 1992, 188, 438–450. [Google Scholar] [CrossRef]
- Carruth, L.M.; Morse, B.A.; Clements, J.E. The leucine domain of the visna virus Tat protein mediates targeting to an AP-1 site in the viral long terminal repeat. J. Virol. 1996, 70, 4338–4344. [Google Scholar] [PubMed]
- Morse, B.A.; Carruth, L.M.; Clements, J.E. Targeting of the visna virus Tat protein to AP-1 sites: Interactions with the bZIP domains of fos and jun in vitro and in vivo. J. Virol. 1999, 73, 37–45. [Google Scholar] [PubMed]
- Barber, S.A.; Bruett, L.; Douglass, B.R.; Herbst, D.S.; Zink, M.C.; Clements, J.E. Visna virus-induced activation of MAPK is required for virus replication and correlates with virus-induced neuropathology. J. Virol. 2002, 76, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.L.; Small, J.; Clements, J.E. Sequences in the visna virus long terminal repeat that control transcriptional activity and respond to viral trans-activation: involvement of AP-1 sites in basal activity and trans-activation. J. Virol. 1989, 63, 3001–3015. [Google Scholar] [PubMed]
- Harmache, A.; Vitu, C.; Russo, P.; Bouyac, M.; Hieblot, C.; Peveri, P.; Vigne, R.; Suzan, M. The caprine arthritis encephalitis virus tat gene is dispensable for efficient viral replication in vitro and in vivo. J. Virol. 1995, 69, 5445–5454. [Google Scholar] [PubMed]
- Murphy, B.G.; Hotzel, I.; Jasmer, D.P.; Davis, W.C.; Knowles, D. TNFalpha and GM-CSF-induced activation of the CAEV promoter is independent of AP-1. Virology 2006, 352, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Sepp, T.; Tong-Starksen, S.E. STAT1 pathway is involved in activation of caprine arthritis-encephalitis virus long terminal repeat in monocytes. J. Virol. 1997, 71, 771–777. [Google Scholar] [PubMed]
- Guo, D.; Dunbar, J.D.; Yang, C.H.; Pfeffer, L.M.; Donner, D.B. Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J. Immunol. 1998, 160, 2742–2750. [Google Scholar] [PubMed]
- Jackson, S.H.; Yu, C.R.; Mahdi, R.M.; Ebong, S.; Egwuagu, C.E. Dendritic cell maturation requires STAT1 and is under feedback regulation by suppressors of cytokine signaling. J. Immunol. 2004, 172, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Del Russo, N.; Stellacci, E.; Testa, U.; Marziali, G.; Battistini, A. STAT1 activation during monocyte to macrophage maturation: role of adhesion molecules. Int. Immunol. 1999, 11, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Reina, R.; Grego, E.; Bertolotti, L.; De Meneghi, D.; Rosati, S. Genome analysis of small-ruminant lentivirus genotype E: a caprine lentivirus with natural deletions of the dUTPase subunit, vpr-like accessory gene, and 70-base-pair repeat of the U3 region. J. Virol. 2009, 83, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.C.; Andresdottir, V.; Fevereiro, M. Cellular specificity and replication rate of Maedi Visna virus in vitro can be controlled by LTR sequences. Arch. Virol. 2005, 150, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Hreggvidsdottir, H.S.; Agnarsdottir, G.; Matthiasdottir, S.; Ogmundsdottir, M.H.; Jonsson, S.R.; Georgsson, G.; Ingvarsson, S.; Andresson, O.S.; Andresdottir, V. Duplicated sequence motif in the long terminal repeat of maedi-visna virus extends cell tropism and is associated with neurovirulence. J. Virol. 2007, 81, 4052–4057. [Google Scholar] [CrossRef] [PubMed]
- Agnarsdottir, G.; Thorsteinsdottir, H.; Oskarsson, T.; Matthiasdottir, S.; Haflidadottir, B.S.; Andresson, O.S.; Andresdottir, V. The long terminal repeat is a determinant of cell tropism of maedi-visna virus. J. Gen. Virol. 2000, 81, 1901–1905. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; McElliott, V.; Vapniarsky, N.; Oliver, A.; Rowe, J. Tissue tropism and promoter sequence variation in caprine arthritis encephalitis virus infected goats. Virus Res. 2010, 151, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Adedeji, A.O.; Barr, B.; Gomez-Lucia, E.; Murphy, B. A polytropic caprine arthritis encephalitis virus promoter isolated from multiple tissues from a sheep with multisystemic lentivirus-associated inflammatory disease. Viruses 2013, 5, 2005–2018. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Hillman, C.; Castillo, D.; Vapniarsky, N.; Rowe, J. The presence or absence of the gamma-activated site determines IFN gamma-mediated transcriptional activation in CAEV promoters cloned from the mammary gland and joint synovium of a single CAEV-infected goat. Virus Res. 2012, 163, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Karn, J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr. HIV Res. 2011, 9, 554–567. [Google Scholar] [PubMed]
- Narlikar, G.J.; Fan, H.-Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002, 108, 475–487. [Google Scholar] [CrossRef]
- Wolffe, A. Nucleosome positioning and modification: chromatin structures that potentiate transcription. Trends Biochem. Sci. 1994, 19, 240–244. [Google Scholar] [CrossRef]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [PubMed]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [PubMed]
- Verdin, E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar] [PubMed]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [PubMed]
- Coull, J.J.; Romerio, F.; Sun, J.-M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The Human Factors YY1 and LSF Repress the Human Immunodeficiency Virus Type 1 Long Terminal Repeat via Recruitment of Histone Deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Okamoto, T. Transcriptional Repression of Human Immunodeficiency Virus Type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Biol. Cell 2002, 22, 2965–2973. [Google Scholar] [CrossRef]
- Williams, S.; Chen, L.; Kwon, C.; Ruiz-Jarabo, E.; Greene, W. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.-R.; Henklein, P.; Schubert, U.; Zhou, M.-M.; Verdin, E.; Ott, M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, T.; Parra, M.; Vries, R.G.J.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar] [CrossRef]
- Tréand, C.; du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [PubMed]
- Agbottah, E.; Deng, L.; Dannenberg, L.O.; Pumfery, A.; Kashanchi, F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology 2006. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA Polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Col, E. The Histone Acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar] [CrossRef] [PubMed]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 Regulates HIV Transcription via Tat Deacetylation. PloS Biol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Sakane, N.; Kwon, H.-S.; Pagans, S.; Kaehlcke, K.; Mizusawa, Y.; Kamada, M.; Lassen, K.G.; Chan, J.; Greene, W.C.; Schnoelzer, M.; et al. Activation of HIV Transcription by the Viral Tat Protein Requires a Demethylation Step Mediated by Lysine-specific Demethylase 1 (LSD1/KDM1). PLoS Pathog. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Dorr, A.; Hetzer-Egger, C.; Kaehlcke, K.; Schnolzer, M.; Henklein, P.; Cole, P.; Zhou, M.-M.; Verdin, E. Tat acetylation: a regulatory switch between early and late phases in HIV transcription elongation. Novartis Found. Symp. 2004, 259, 182–193. [Google Scholar] [PubMed]
- Pagans, S.; Kauder, S.E.; Kaehlcke, K.; Sakane, N.; Schroeder, S.; Dormeyer, W.; Trievel, R.C.; Verdin, E.; Schnolzer, M.; Ott, M. The Cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription. Cell. Host Microbe 2010, 7, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Lusic, M.; Cereseto, A.; Giacca, M. Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Mol. Biol. Cell. 2008, 28, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Cho, W.-K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Enderle, D.; Beisel, C.; Stadler, M.B.; Gerstung, M.; Athri, P.; Paro, R. Polycomb preferentially targets stalled promoters of coding and noncoding transcripts. Genome Res. 2011, 21, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Herbein, G. Molecular Understanding of HIV-1 Latency. Adv. Virol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Groen, J.; Morris, K. Chromatin, Non-Coding RNAs, and the Expression of HIV. Viruses 2013, 5, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Winckelmann, A.; Ostergaard, L.; Sogaard, O.S. Eliminating the latent HIV reservoir by reactivation strategies: Advancing to clinical trials. Hum. Vaccin. Immunother. 2013, 9, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.M.; Burch, B.D.; Soriano-Sarabia, N.; Margolis, D.M. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Blankson, J.N.; Siliciano, R.F. Developing strategies for HIV-1 eradication. Trends Immuno.l 2012, 33, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Sereti, I.; Tambussi, G.; Routy, J.P.; Lelievre, J.D.; Delfraissy, J.F.; Molina, J.M.; Fischl, M.; Goujard, C.; Rodriguez, B.; et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: Results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin. Infect. Dis. 2012, 55, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Strong, C.L.; Guerra, H.P.; Mathew, K.R.; Roy, N.; Simpson, L.R.; Schiller, M.R. Damaging the integrated HIV proviral DNA with TALENs. PloS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Mariyanna, L.; Priyadarshini, P.; Hofmann-Sieber, H.; Krepstakies, M.; Walz, N.; Grundhoff, A.; Buchholz, F.; Hildt, E.; Hauber, J. Excision of HIV-1 proviral DNA by recombinant cell permeable tre-recombinase. PloS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Hauber, I.; Hauber, J.; Buchholz, F. HIV-1 proviral DNA excision using an evolved recombinase. Science 2007, 316, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- June, C. Introduction of Acquired CCR5 Deficiency with Zinc Finger Nuclease-Modified Autologous CD4 T Cells (SB-728-T) Correlates with Increases in CD4 Count and Effects on Viral Load in HIV-Infected Subjects. In Proceedings of 19th Conference on Retroviruses and Opportunistic Infections, Seattle, USA, 5–8 March 2012.
- Lalezari, J. A Single Infusion of Zinc Finger Nuclease CCR5 Modified Autologous CD4 T Cells (SB-728-T) Increases CD4 Counts and Leads to Decrease in HIV Proviral Load in an Aviremic HIV-Infected Subject. In Proceedings of 19th Conference on Retroviruses and Opportunistic Infections, Seattle, USA, 5–8 March 2012.
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Ganepola, S. Eradication of HIV by transplantation of CCR5-deficient hematopoietic stem cells. ScientificWorldJournal 2011, 11, 1068–1076. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Mens, H.; Kearney, M.; Wiegand, A.; Shao, W.; Schonning, K.; Gerstoft, J.; Obel, N.; Maldarelli, F.; Mellors, J.W.; Benfield, T.; et al. HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J. Virol. 2010, 84, 12971–12981. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.A.; Brennan, T.P.; Bailey, J.R.; Ray, S.C.; Siliciano, R.F.; Blankson, J.N. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J. Virol. 2010, 84, 7018–7028. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Vasir, B.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hu, Z.; Li, J.Z.; Sciaranghella, G.; Busch, M.P.; Keating, S.M.; Gallien, S.; Lin, N.H.; Giguel, F.F.; Lavoie, L.; et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 2013, 207, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Hanhauser, E.; Sirignano, M.N.; Li, J.Z.; Lichterfeld, M.; Marty, F.M.; Armand, P.; Soiffer, R.J.; Altfeld, M.; Kuritzkes, D.R. HIV-1 Rebound Following Allogeneic Stem Cell Transplantation and Treatment Interruption. In Proceeding of 21st Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 3–6 March 2014.
- Frange, P.; Avettand-Fenoel, A.F.V.; Bellaton, E.; Deschamps, D.; Angin, M.; Caillat-Zucman, S.G.; Peytavin, J.; Chenadec, L.; Warszawski, J.; Rouzioux, C.; Saez-Cirion, A. HIV-1 Virological Remission for More Than 11 Years after Interruption of Early Initiated Antiretroviral Therapy in A Perinatally-Infected Child. In Proceedings of IAS 2015—8th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Vancouver, Canada, 22 July 2015.
- Persaud, D.; Gay, H.; Ziemniak, C.; Chen, Y.H.; Piatak, M., Jr.; Chun, T.W.; Strain, M.; Richman, D.; Luzuriaga, K. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N. Engl. J. Med. 2013, 369, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.; Barre-Sinoussi, F.; Nugeyre, M. Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science 1984, 225, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.; Champagne, E.; Chamaret, S.; Gruest, J.; Guetard, D.; Hercend, T.; Gluckman, J.C.; Montagnier, L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 1984, 312, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.D.; Rota, T.; Hirsch, M. Infection of monocyte/macrophages by human T lymphotropic virus type III. J. Clin. Invest. 1986, 77, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Swiggard, W.J.; Baytop, C.; Yu, J.J.; Dai, J.; Li, C.; Schretzenmair, R.; Theodosopoulos, T.; O’Doherty, U. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J. Virol. 2005, 79, 14179–14188. [Google Scholar] [CrossRef] [PubMed]
- Chanel, C.; Staropoli, I.; Baleux, F.; Amara, A.; Valenzuela-Fernandez, A.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Altmeyer, R. Low levels of co-receptor CCR5 are sufficient to permit HIV envelope-mediated fusion with resting CD4 T cells. AIDS 2002, 16, 2337–2340. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, D.A.; Penn, M.L.; Korin, Y.D.; Scripture-Adams, D.D.; Zack, J.A.; Kreisberg, J.F.; Roederer, M.; Sherman, M.P.; Chin, P.S.; Goldsmith, M.A. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 2001, 15, 671–682. [Google Scholar] [CrossRef]
- Chu, T.; Carruth, L.M.; Finzi, D. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Balnkson, J.; Siliciano, J. Latent infection of CD4+T cells provides a mechanism for life long persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [PubMed]
- Siliciano, J.; Siliciano, R.F. Latency and viral persistence in HIV-1 infection. J. Clin. Invest. 2000, 106, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Blankson, J.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef]
- Williams, K.C.; Hickey, W.F. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu. Rev. Neurosci. 2002, 25, 537–562. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 2002, 40, 240–251. [Google Scholar] [CrossRef]
- Smith, P.D.; Meng, G.; Salazar-Gonzalez, J.F.; Shaw, G.M. Macrophage HIV-1 infection and the gastrointestinal tract reservoir. J. Leukoc. Biol. 2003, 74, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Meng, G.; Ochsenbauer, C.; Clapham, P.R.; Grams, J.; Novak, L.; Kappes, J.C.; Smythies, L.E.; Smith, P.D. Stromal down-regulation of macrophage CD4/CCR5 expression and NF-κB activation mediates HIV-1 non-permissiveness in intestinal macrophages. PLoS Pathog. 2011. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of HIV-1 from the Macrophage Reservoir: An Uncertain Goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Saksena, N.K.; Wang, B.; Zhou, L.; Soedjono, M.; Ho, Y.S.; Conceicao, V. HIV reservoirs in vivo and new strategies for possible eradication of HIV from the reservoir sites. HIV/AIDS 2010, 2, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.T.; Henry, K.; Zupancic, M. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 1996, 274, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Bertolotti, L.; Juganaru, M.; Glaria, I.; de Andres, D.; Amorena, B.; Rosati, S.; Reina, R. Small ruminant macrophage polarization may play a pivotal role on lentiviral infection. Vet. Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.R.; Roche, M.; Flynn, J.K.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. Is the central nervous system a reservoir of HIV-1? Curr. Opin. HIV AIDS 2014, 9, 552–558. [Google Scholar] [CrossRef]
- Churchill, M.; Nath, A. Where does HIV hide? A focus on the central nervous system. Curr. Opin. HIV AIDS 2013, 8, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Veenstra, M.; Gaskill, P.J.; Morgello, S.; Calderon, T.M.; Berman, J.W. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr. HIV Res. 2014, 12, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.; Major, E. HIV-1 and the brain: connections between HIV-1-associated dementia, neuropathology and neuroimmunology. Microb. Infect. 2002, 4, 301–308. [Google Scholar] [CrossRef]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain. Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Benavides, J.; Garcia-Pariente, C.; Fuertes, M.; Ferreras, M.C.; Garcia-Marin, J.F.; Juste, R.A.; Perez, V. Maedi-visna: the meningoencephalitis in naturally occurring cases. J. Comp. Pathol. 2009, 140, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Benavides, J.; Fuertes, M.; Garcia-Pariente, C.; Ferreras, M.C.; Garcia Marin, J.F.; Perez, V. Natural cases of visna in sheep with myelitis as the sole lesion in the central nervous system. J. Comp. Pathol. 2006, 134, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Gianella, S.; Sinclair, E.; Epling, L.; Li, Q.; Duan, L.; Choi, A.L.; Girling, V.; Ho, T.; Li, P.; et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J. Infect. Dis. 2010, 202, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Cerf-Bensussan, N.; Guy-Grand, D. Intestinal intraepithelial lymphocytes. Gastroenterol. Clin. North. Am. 1991, 20, 549–576. [Google Scholar] [PubMed]
- Schneider, T.; Jahn, H.U.; Schmidt, W.; Riecken, E.O.; Zeitz, M.; Ullrich, R. Loss of CD4 T lymphocytes in patients infected with human immunodeficiency virus type 1 is more pronounced in the duodenal mucosa than in the peripheral blood. Berlin diarrhea/wasting syndrome study group. Gut 1995, 37, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Olech, M.; Kubis, P.; Lipecka, C.; Junkuszew, A.; Gruszecki, T.M.; Kuźmak, J. Presence of specific antibodies and proviral DNA of small ruminant lentiviruses in lambs in their first weeks of life. Bull. Vet. Inst. Pulawy 2014, 58, 507–511. [Google Scholar] [CrossRef]
- Grossi, P.; Giudice, C.; Bertoletti, I.; Cioccarelli, G.; Brocchi, E.; Cammarata, G.; Gelmetti, D. Immunohistochemical detection of the p27 capsid protein of caprine arthritis-encephalitis virus (CAEV) in bone-marrow cells of seropositive goats. J. Comp. Pathol. 2005, 133, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Ravazzolo, A.P.; Nenci, C.; Vogt, H.-R.; Waldvogel, A.; Obexer-Ruff, G.; Peterhans, E.; Bertoni, G. Viral load, organ distribution, histopathological lesions, and cytokine mRNA expression in goats infected with a molecular clone of the caprine arthritis encephalitis virus. Virology 2006, 350, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Lambert-Niclot, S.; Tubiana, R.; Beaudoux, C.; Lefebvre, G.; Caby, F.; Bonmarchand, M.; Naouri, M.; Schubert, B.; Dommergues, M.; Calvez, V.; et al. Detection of HIV-1 RNA in seminal plasma samples from treated patients with undetectable HIV-1 RNA in blood plasma on a 2002–2011 survey. AIDS 2012, 26, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, A.G.; Tubiana, R.; Lambert-Niclot, S.; Lefebvre, G.; Dominguez, S.; Bonmarchand, M.; Vauthier-Brouzes, D.; Marguet, F.; Mousset-Simeon, N.; Peytavin, G.; et al. Detection of HIV-1 RNA in seminal plasma samples from treated patients with undetectable HIV-1 RNA in blood plasma. AIDS 2008, 22, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Van Heeswijk, R.; Hoetelmans, R. Concentration of nevirapine, lamivudine and stavudine in semen of HIV-1 infected men. AIDS 2000, 1979–1984. [Google Scholar] [CrossRef]
- Sheth, P.M.; Kovacs, C.; Kemal, K.S.; Jones, R.B.; Raboud, J.M.; Pilon, R.; la Porte, C.; Ostrowski, M.; Loutfy, M.; Burger, H.; et al. Persistent HIV RNA shedding in semen despite effective antiretroviral therapy. AIDS 2009, 23, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Le Tortorec, A.; Dejucq-Rainsford, N. HIV infection of the male genital tract—consequences for sexual transmission and reproduction. Int. J. Androl. 2010, 33, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Dejucq-Rainsford, N.; Jegou, B. Viruses in semen and male genital tissues--consequences for the reproductive system and therapeutic perspectives. Curr. Pharm. Des. 2004, 10, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Lasheeb, A.S.; King, J.; Ball, J.K.; Curran, R.; Barratt, C.L.; Afnan, M.; Pillay, D. Semen characteristics in HIV-1 positive men and the effect of semen washing. Genitourin. Med. 1997, 73, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Pudney, J.; Anderson, D. Orchitis and human immunodeficiency virus type 1 infected cells in reproductive tissues from men with the acquired immune deficiency syndrome. Am. J. Pathol. 1991, 139, 149–160. [Google Scholar] [PubMed]
- Muciaccia, B.; Filippini, A.; Ziparo, E.; Colelli, F.; Baroni, C.D.; Stefanini, M. Testicular germ cells of HIV-seropositive asymptomatic men are infected by the virus. J. Reprod. Immunol. 1998, 41, 81–93. [Google Scholar] [CrossRef]
- Muciaccia, B.; Corallini, S.; Vicini, E.; Padula, F.; Gandini, L.; Liuzzi, G.; Lenzi, A.; Stefanini, M. HIV-1 viral DNA is present in ejaculated abnormal spermatozoa of seropositive subjects. Hum. Reprod. 2007, 22, 2868–2878. [Google Scholar] [CrossRef] [PubMed]
- Galvin, S.R.; Cohen, M.S. Genital tract reservoirs. Curr. Opin. HIV AIDS 2006, 1, 162–166. [Google Scholar] [PubMed]
- Bull, M.E.; Learn, G.H.; McElhone, S.; Hitti, J.; Lockhart, D.; Holte, S.; Dragavon, J.; Coombs, R.W.; Mullins, J.I.; Frenkel, L.M. Monotypic human immunodeficiency virus type 1 genotypes across the uterine cervix and in blood suggest proliferation of cells with provirus. J. Virol. 2009, 83, 6020–6028. [Google Scholar] [CrossRef]
- Si-Mohamed, A.; Kazatchkine, M.D.; Heard, I.; Goujon, C.; Prazuck, T.; Aymard, G.; Cessot, G.; Kuo, Y.H.; Bernard, M.C.; Diquet, B.; et al. Selection of drug-resistant variants in the female genital tract of human immunodeficiency virus type 1-infected women receiving antiretroviral therapy. J. Infect. Dis. 2000, 182, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Ali Al Ahmad, M.Z.; Dubreil, L.; Chatagnon, G.; Khayli, Z.; Theret, M.; Martignat, L.; Chebloune, Y.; Fieni, F. Goat uterine epithelial cells are susceptible to infection with Caprine Arthritis Encephalitis Virus (CAEV) in vivo. Vet. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lamara, A.; Fieni, F.; Mselli-Lakhal, L.; Tainturier, D.; Chebloune, Y. Efficient replication of caprine arthritis-encephalitis virus in goat granulosa cells. Virus Res. 2001, 79, 165–172. [Google Scholar] [CrossRef]
- Lamara, A.; Fieni, F.; Mselli-Lakhal, L.; Tainturier, D.; Chebloune, Y. Epithelial cells from goat oviduct are highly permissive for productive infection with caprine arthritis-encephalitis virus (CAEV). Virus Res. 2002, 87, 69–77. [Google Scholar] [CrossRef]
- Ali Al Ahmad, M.Z.; Fieni, F.; Guiguen, F.; Larrat, M.; Pellerin, J.L.; Roux, C.; Chebloune, Y. Cultured early goat embryos and cells are susceptible to infection with caprine encephalitis virus. Virology 2006, 353, 307–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lamara, A.; Fieni, F.; Chatagnon, G.; Larrat, M.; Dubreil, L.; Chebloune, Y. Caprine arthritis encephalitis virus (CAEV) replicates productively in cultured epididymal cells from goats. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 397–404. [Google Scholar] [CrossRef]
- Ali Al Ahmad, M.Z.; Fieni, F.; Pellerin, J.L.; Guiguen, F.; Cherel, Y.; Chatagnon, G.; Bouzar, A.B.; Chebloune, Y. Detection of viral genomes of caprine arthritis-encephalitis virus (CAEV) in semen and in genital tract tissues of male goat. Theriogenology 2008, 69, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Turchetti, A.P.; Paniago, J.J.; da Costa, L.F.; da Cruz, J.C.; Braz, G.F.; Gouveia, A.M.; Paixao, T.A.; Santos, R.L.; Heinemann, M.B. Distribution of caprine arthritis encephalitis virus provirus, RNA, and antigen in the reproductive tract of one naturally and seven experimentally infected bucks. Theriogenology 2013, 80, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Fieni, F.; Pellerin, J.L.; Roux, C.; Poulin, N.; Baril, G.; Fatet, A.; Valas, S.; Chatagnon, G.; Mermillod, P.; Guignot, F. Can caprine arthritis encephalitis virus (CAEV) be transmitted by in vitro fertilization with experimentally infected sperm? Theriogenology 2012, 77, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Alimohammadi, A.; Coker, R.; Miller, R.; Mitchell, D.; Williamson, J.; Clarke, J. Genotypic variants of HIV-1 from peripheral blood and lungs of AIDS patients. AIDS 1997, 11, 831–832. [Google Scholar] [PubMed]
- White, N.; Israel-Biet, D.; coker, R.; Mitchell, D.; Weber, J.; Clarke, J. Different resistance mutations can be detected simultaneously in the blood and the lung of HIV-1 infected individuals on antiretroviral therapy. J. Med. Virol. 2004, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retroviruses 2015, 31, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Almodovar, S. The complexity of HIV persistence and pathogenesis in the lung under antiretroviral therapy: challenges beyond AIDS. Viral Immunol. 2014, 27, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Costiniuk, C.T.; Jenabian, M.A. The lungs as anatomical reservoirs of HIV infection. Rev. Med. Virol. 2014, 24, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, L.; Ross, M.; Tanji, N. Renal epithelium is a previously unrecognized site of HIV-1 infection. J. Am. Soc. Nephrol. 2000, 2079–2087. [Google Scholar]
- Marras, D.; Bruggeman, L.; Gao, F. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat. Med. 2002, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Blasi, M.; Balakumaran, B.; Chen, P.; Negri, D.R.; Cara, A.; Chen, B.K.; Klotman, M.E. Renal epithelial cells produce and spread HIV-1 via T-cell contact. AIDS 2014, 28, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Dejucq-Rainsford, N.; Avettand-Fenoel, V.; Viard, J.P.; Anglicheau, D.; Bienaime, F.; Muorah, M.; Galmiche, L.; Gribouval, O.; Noel, L.H.; et al. The kidney as a reservoir for HIV-1 after renal transplantation. J. Am. Soc. Nephrol. 2014, 25, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.G.; Barin, B.; Hatano, H.; Rogers, R.L.; Roland, M.E.; Lee, T.H.; Busch, M.; Deeks, S.G. Reduction of HIV persistence following transplantation in HIV-infected kidney transplant recipients. Am. J. Transplant. 2014, 14, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- McNeilly, T.N.; Baker, A.; Brown, J.K.; Collie, D.; Maclachlan, G.; Rhind, S.M.; Harkiss, G.D. Role of alveolar macrophages in respiratory transmission of visna/maedi virus. J. Virol. 2008, 82, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- McNeilly, T.N.; Tennant, P.; Lujan, L.; Perez, M.; Harkiss, G.D. Differential infection efficiencies of peripheral lung and tracheal tissues in sheep infected with Visna/maedi virus via the respiratory tract. J. Gen. Virol. 2007, 88, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.; Watt, N.; Hopkins, J.; Harkiss, G.; Woodall, C. Quantitative analysis of Maedi Visna virus DNA load in peripheral blood monocytes and alveolar macrophages. J. Virol. Methods 2000, 13–20. [Google Scholar] [CrossRef]
- Watt, N.J.; MacIntyre, N.; Collie, D.; Sargan, D.; McConnell, I. Phenotypic analysis of lymphocyte populations in the lungs and regional lymphoid tissue of sheep naturally infected with maedi visna virus. Clin. Exp. Immunol. 1992, 90, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Patton, K.M.; Bildfell, R.J.; Anderson, M.L.; Cebra, C.K.; Valentine, B.A. Fatal Caprine arthritis encephalitis virus-like infection in 4 Rocky Mountain goats (Oreamnos americanus). J. Vet. Diagn. Invest. 2012, 24, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Arrode-Brusés, G.; Hegde, R.; Jin, Y.; Liu, Z.; Narayan, O.; Chebloune, Y. Immunogenicity of a lentiviral-based DNA vaccine driven by the 5′LTR of the naturally attenuated caprine arthritis encephalitis virus (CAEV) in mice and macaques. Vaccine 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Chebloune, Y.; Moussa, M.; Arrode-Bruses, G.; Gagnon, J. Cowpox helped against smallpox; will the goat lentivirus (Caprine Arthritis Encephalitis Virus) help against HIV-1? AIDS Res. Hum. Retroviruses 2015. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bose, D.; Gagnon, J.; Chebloune, Y. Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses. Vet. Sci. 2015, 2, 293-348. https://doi.org/10.3390/vetsci2040293
Bose D, Gagnon J, Chebloune Y. Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses. Veterinary Sciences. 2015; 2(4):293-348. https://doi.org/10.3390/vetsci2040293
Chicago/Turabian StyleBose, Deepanwita, Jean Gagnon, and Yahia Chebloune. 2015. "Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses" Veterinary Sciences 2, no. 4: 293-348. https://doi.org/10.3390/vetsci2040293
APA StyleBose, D., Gagnon, J., & Chebloune, Y. (2015). Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses. Veterinary Sciences, 2(4), 293-348. https://doi.org/10.3390/vetsci2040293