

In Silico Designed Multi-Epitope Vaccine Based on the Conserved Fragments in Viral Proteins for Broad-Spectrum Protection Against Porcine Reproductive and Respiratory Syndrome Virus


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of PRRSV Protein Sequence
2.2. Step-by-Step Selection of Epitopes
2.3. Design and Assembly of Candidate Vaccines
2.4. Evaluation of Candidate Vaccines in Physicochemical and Immune Properties
2.5. Prediction and Validation of Molecular Structure for Candidate Vaccines
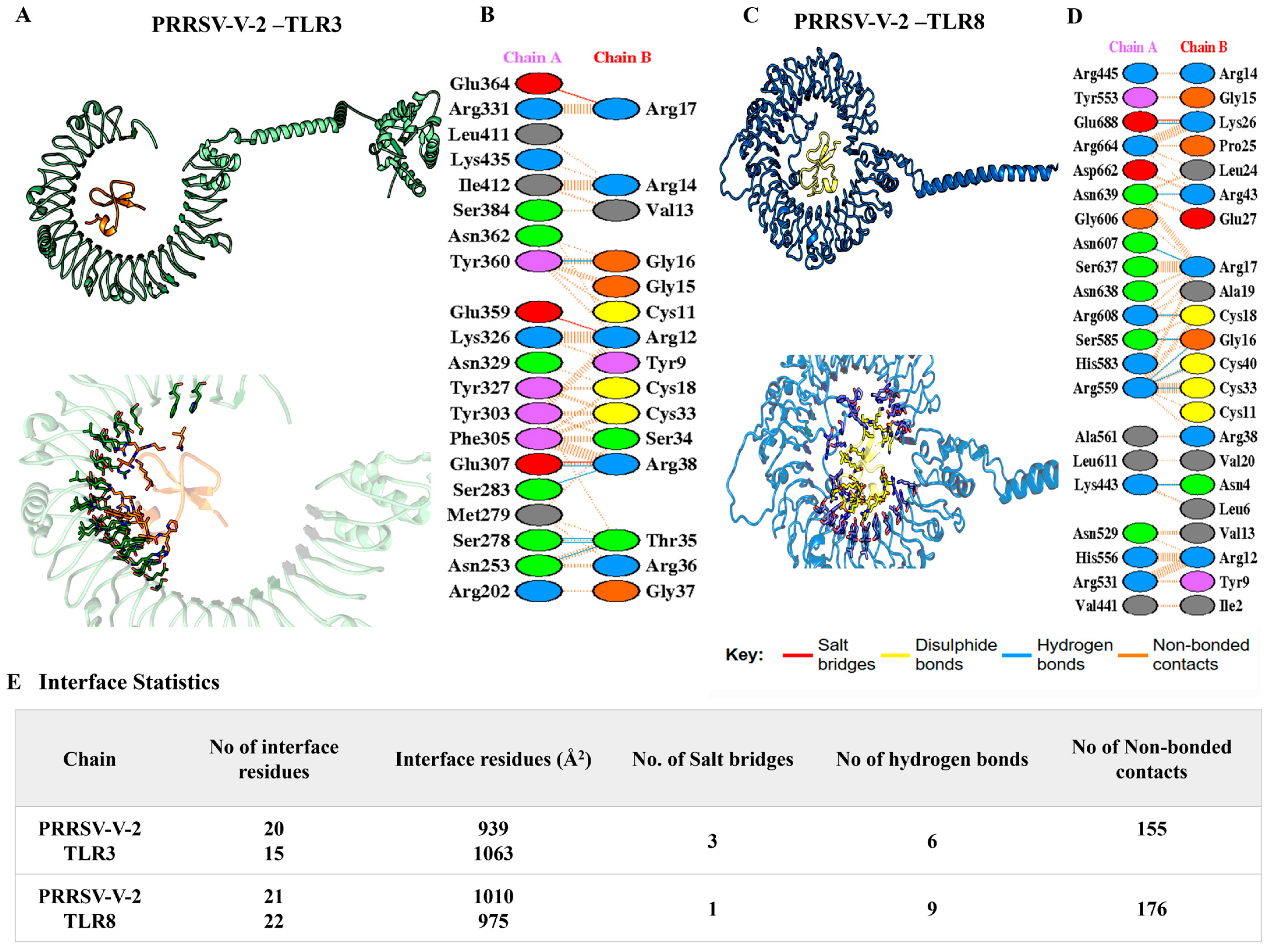
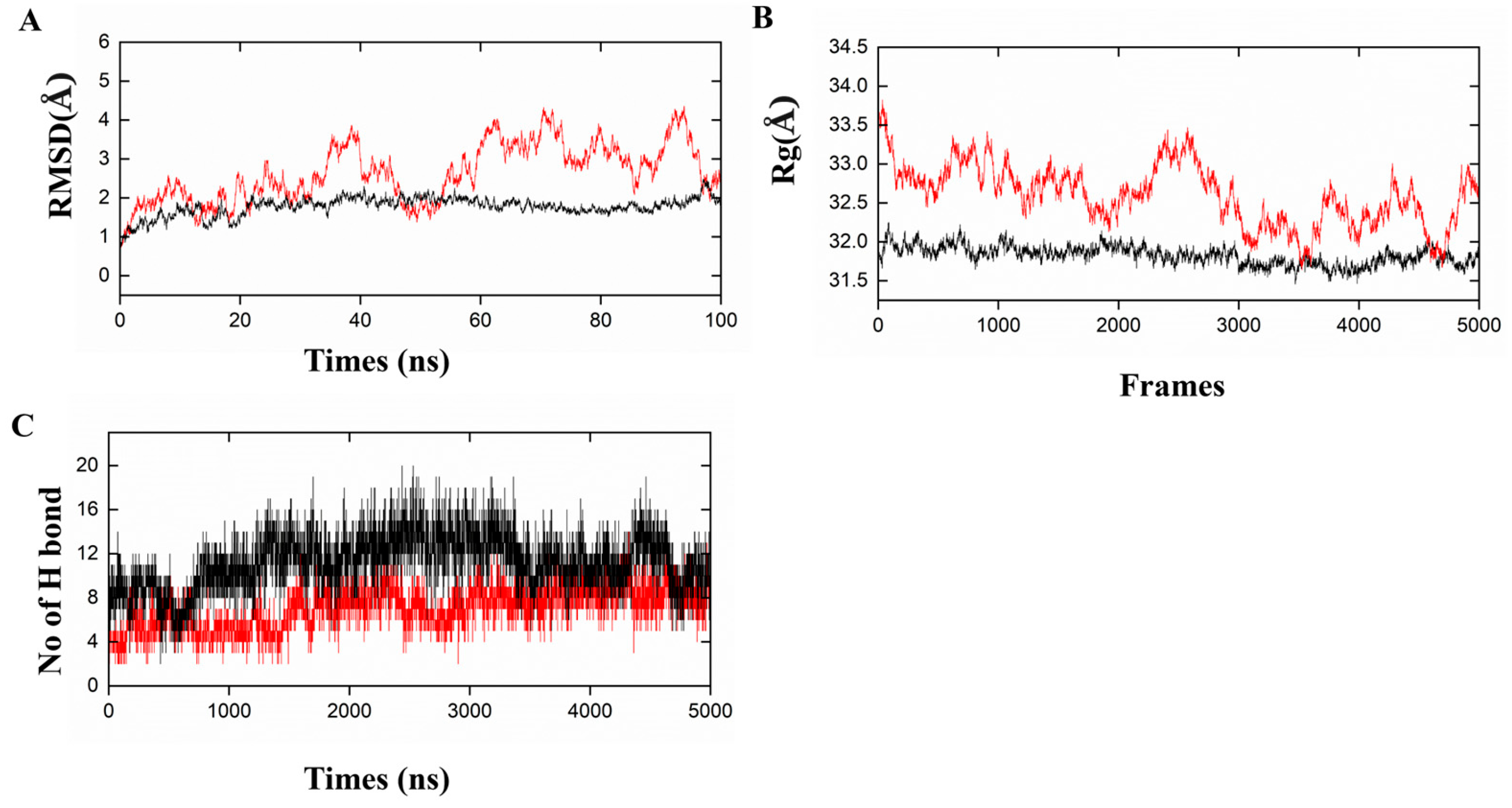
2.6. Molecular Docking and Dynamic Analysis of Vaccine-TLR (Toll-like Receptors) Complexes
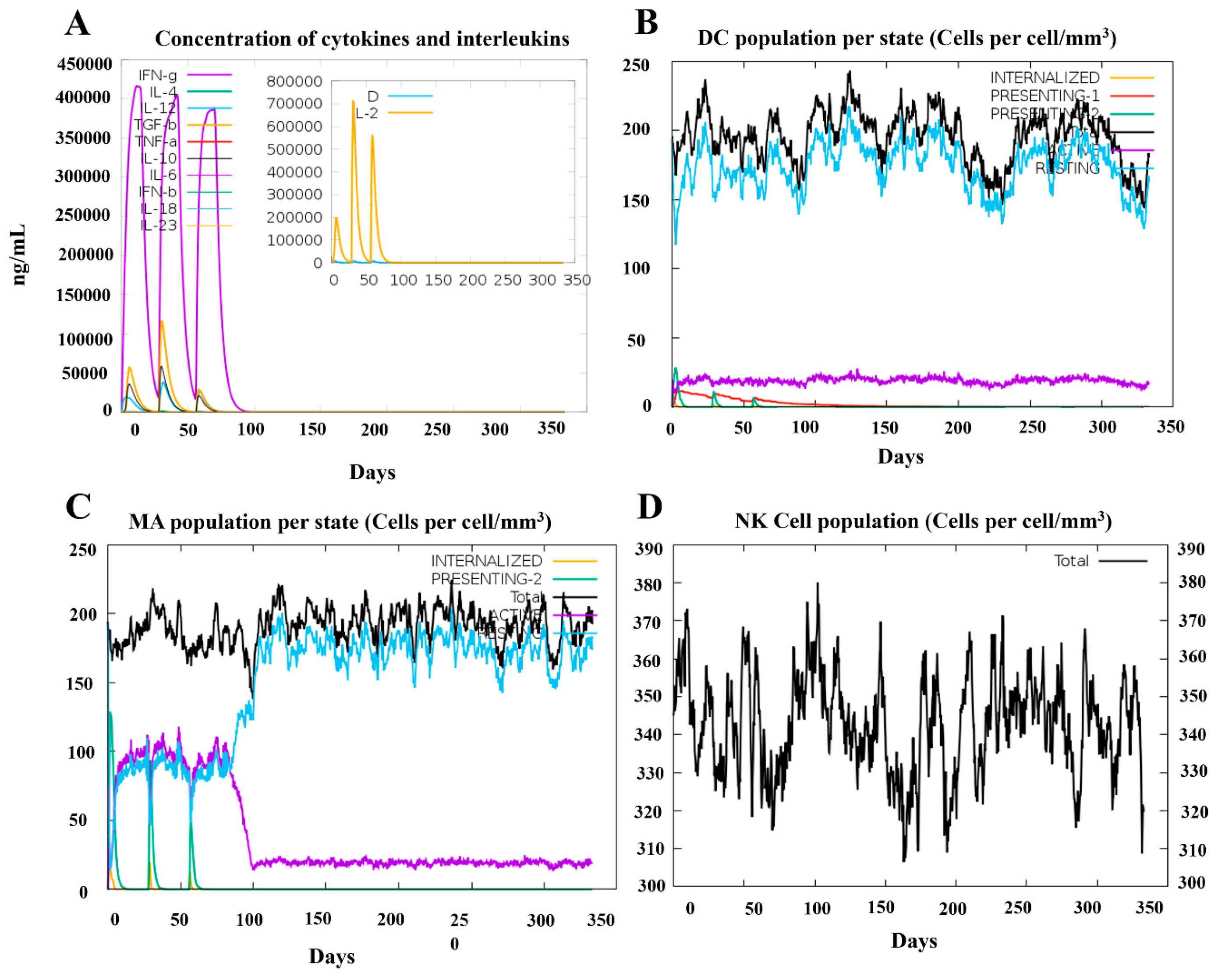
2.7. Immune Simulation of Candidate Vaccines
2.8. In Silico Cloning of Selected Vaccine Candidate
3. Results
3.1. Extraction of Conserved Fragments from PRRSV Protein Sequences Collected Worldwide
3.2. Prediction and Evaluation of Epitopes for B Cells, CTLs, and HTLs
3.3. Design of Three Candidate Vaccines
- PRRSV-V-1: Core antigen attached with S50 L7/12 ribosomal protein on N-terminal.
- PRRSV-V-2: Core antigen attached with β-defensin on the N-terminal.
- PRRSV-V-3: Core antigen attached with HBHA adjuvant on N-terminal.
3.4. Properties of Candidate Vaccines in Physicochemical and Immunology
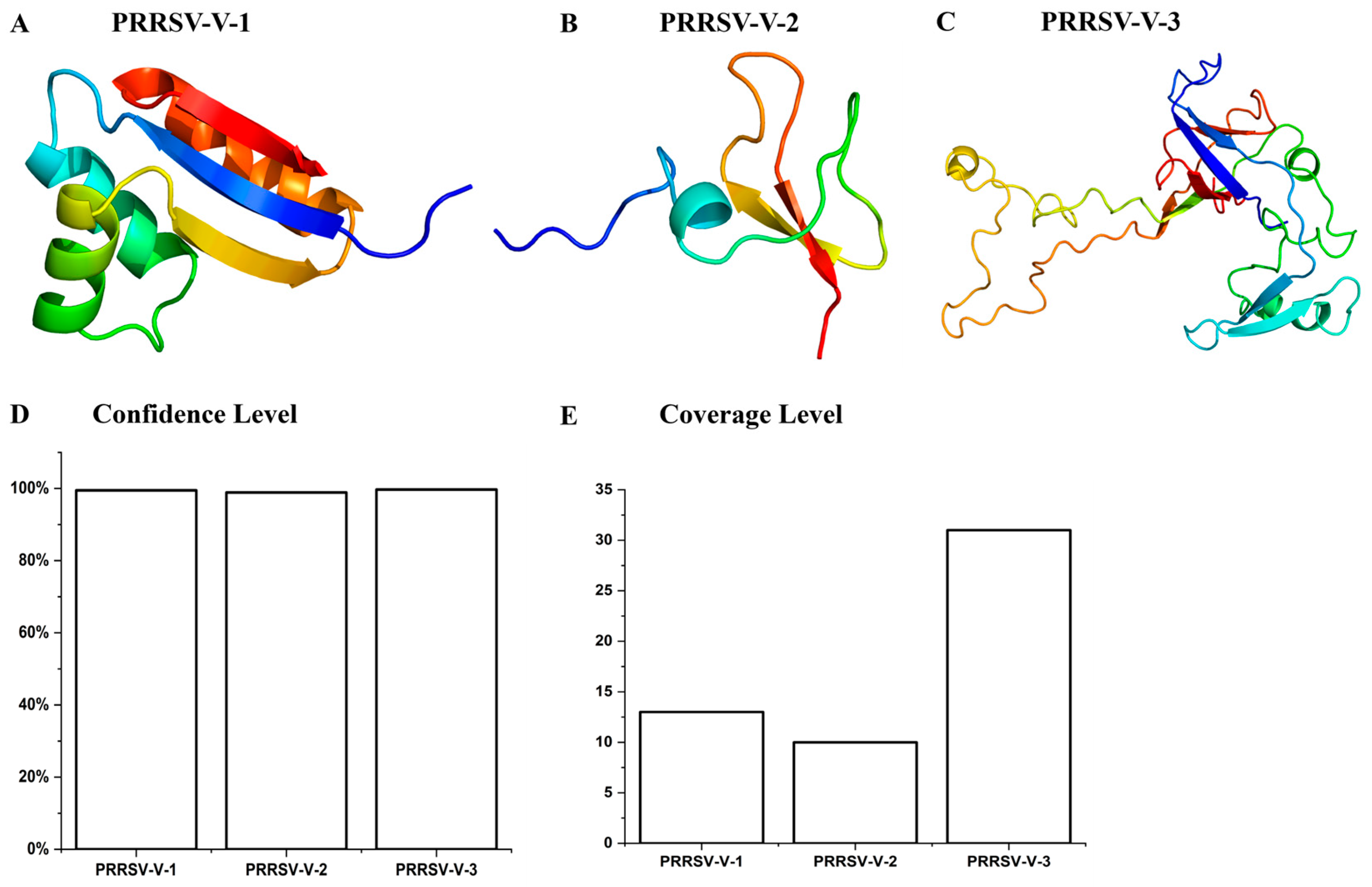
3.5. Prediction and Refinement of Secondary and Tertiary Structure
3.6. Molecular Docking and Dynamics Simulation for Complexes of Vaccine-Immune Receptor
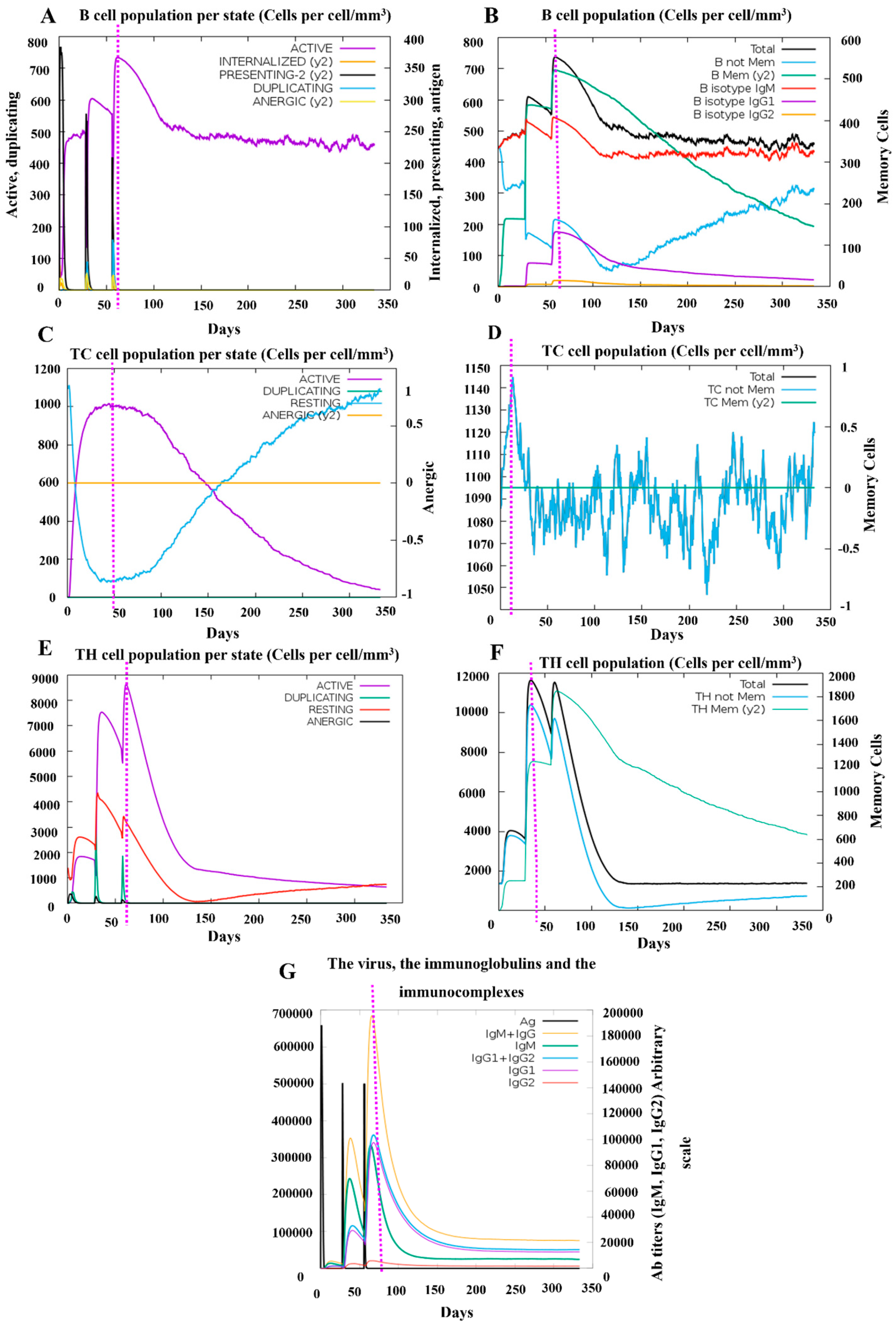
3.7. Simulation of Immune Responses Post-Vaccination
3.8. Codon Optimization of the Final Vaccine Candidate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PRRSV | Porcine respiratory and reproductive syndrome virus |
| PRRSV-V | Porcine respiratory and reproductive syndrome virus-vaccine |
| SLA | Swine Leukocyte Antigen |
| HLA | Human Leukocyte Antigen |
References
- Albina, E. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): An overview. Vet. Microbiol. 1997, 55, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, C.; Provost, C.; Gagnon, C.A. Whole-genome sequencing of porcine reproductive and respiratory syndrome virus from field clinical samples improves the genomic surveillance of the virus. J. Clin. Microbiol. 2020, 58, 10–1128. [Google Scholar] [CrossRef]
- Neumann, E.J.; Kliebenstein, J.B.; Johnson, C.D.; Mabry, J.W.; Bush, E.J.; Seitzinger, A.H.; Green, A.L.; Zimmerman, J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J. Am. Vet. Med. Assoc. 2005, 227, 385–392. [Google Scholar] [CrossRef]
- Renken, C.; Nathues, C.; Swam, H.; Fiebig, K.; Weiss, C.; Eddicks, M.; Ritzmann, M.; Nathues, H. Application of an economic calculator to determine the cost of porcine reproductive and respiratory syndrome at farm-level in 21 pig herds in Germany. Porc. Health Manag. 2021, 7, 3. [Google Scholar] [CrossRef]
- Sun, Q.; Xu, H.; Li, C.; Gong, B.; Li, Z.; Tian, Z.-J.; Zhang, H. Emergence of a novel PRRSV-1 strain in mainland China: A recombinant strain derived from the two commercial modified live viruses Amervac and DV. Front. Vet. Sci. 2022, 9, 974743. [Google Scholar] [CrossRef]
- Yu, F.; Liu, L.; Tian, X.; Chen, L.; Huang, X.; Sun, Y.; Yan, Y.; Tian, Z.; Cai, X.; Liu, D. Genomic analysis of porcine reproductive and respiratory syndrome virus 1 revealed extensive recombination and potential introduction events in China. Vet. Sci. 2022, 9, 450. [Google Scholar] [CrossRef]
- Yu, F.; Yan, Y.; Shi, M.; Liu, H.-Z.; Zhang, H.-L.; Yang, Y.-B.; Huang, X.-Y.; Gauger, P.C.; Zhang, J.; Zhang, Y.-H. Phylogenetics, genomic recombination, and NSP2 polymorphic patterns of porcine reproductive and respiratory syndrome virus in China and the United States in 2014–2018. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Wang, L.-j.; Wan, B.; Guo, Z.; Qiao, S.; Li, R.; Xie, S.; Chen, X.-x.; Zhang, G. Genomic analysis of a recombinant NADC30-like porcine reproductive and respiratory syndrome virus in China. Virus Genes 2018, 54, 86–97. [Google Scholar] [CrossRef]
- Zhao, K.; Ye, C.; Chang, X.-B.; Jiang, C.-G.; Wang, S.-J.; Cai, X.-H.; Tong, G.-Z.; Tian, Z.-J.; Shi, M.; An, T.-Q. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J. Virol. 2015, 89, 10712–10716. [Google Scholar] [CrossRef]
- Ruedas-Torres, I.; Rodríguez-Gómez, I.; Sánchez-Carvajal, J.M.; Larenas-Muñoz, F.; Pallarés, F.; Carrasco, L.; Gómez-Laguna, J. The jigsaw of PRRSV virulence. Vet. Microbiol. 2021, 260, 109168. [Google Scholar] [CrossRef]
- Fang, Y.; Snijder, E.J. The PRRSV replicase: Exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010, 154, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Dea, S.; Gagnon, C.; Mardassi, H.; Pirzadeh, B.; Rogan, D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: Comparison of the North American and European isolates. Arch. Virol. 2000, 145, 659–688. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Gu, G.; Sun, W.; Zhang, Y.-J.; Zhou, E.-M. Improved vaccine against PRRSV: Current progress and future perspective. Front. Microbiol. 2017, 8, 1635. [Google Scholar] [CrossRef]
- Guan, Z.; Pang, L.; Ouyang, Y.; Jiang, Y.; Zhang, J.; Qiu, Y.; Li, Z.; Li, B.; Liu, K.; Shao, D. Secondary Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus (HP-PRRSV2) Infection Augments Inflammatory Responses, Clinical Outcomes, and Pathogen Load in Glaesserella-parasuis-Infected Piglets. Vet. Sci. 2023, 10, 365. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Q.; He, Y.; Zheng, Y.; Sha, H.; Li, G.; Kong, W.; Liao, J.; Zhao, M. Research Progress on the Development of Porcine Reproductive and Respiratory Syndrome Vaccines. Vet. Sci. 2023, 10, 491. [Google Scholar] [CrossRef]
- Wang, H.; Feng, W. Current Status of Porcine Reproductive and Respiratory Syndrome Vaccines. Vaccines 2024, 12, 1387. [Google Scholar] [CrossRef]
- Charerntantanakul, W. Porcine reproductive and respiratory syndrome virus vaccines: Immunogenicity, efficacy and safety aspects. World J. Virol. 2012, 1, 23. [Google Scholar] [CrossRef]
- Renukaradhya, G.J.; Meng, X.-J.; Calvert, J.G.; Roof, M.; Lager, K.M. Inactivated and subunit vaccines against porcine reproductive and respiratory syndrome: Current status and future direction. Vaccine 2015, 33, 3065–3072. [Google Scholar] [CrossRef]
- Chae, C. Commercial PRRS modified-live virus vaccines. Vaccines 2021, 9, 185. [Google Scholar] [CrossRef]
- Lee, M.-A.; Jayaramaiah, U.; You, S.-H.; Shin, E.-G.; Song, S.-M.; Ju, L.; Kang, S.-J.; Hyun, B.-H.; Lee, H.-S. Molecular characterization of porcine reproductive and respiratory syndrome virus in Korea from 2018 to 2022. Pathogens 2023, 12, 757. [Google Scholar] [CrossRef]
- Papatsiros, V. Impact of a killed PRRSV vaccine on sow longevity in a PRRSV infected swine herd. J. Appl. Anim. Res. 2012, 40, 297–304. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.K.; Jung, J.H.; Choi, Y.J.; Kim, J.; Um, C.G.; Hyun, S.B.; Shin, S.; Lee, B.; Jang, G. The assessment of efficacy of porcine reproductive respiratory syndrome virus inactivated vaccine based on the viral quantity and inactivation methods. Virol. J. 2011, 8, 323. [Google Scholar] [CrossRef]
- Van Nieuwstadt, A.; Meulenberg, J.; van Essen-Zanbergen, A.; Petersen-den Besten, A.; Bende, R.; Moormann, R.; Wensvoort, G. Proteins encoded by open reading frames 3 and 4 of the genome of Lelystad virus (Arteriviridae) are structural proteins of the virion. J. Virol. 1996, 70, 4767–4772. [Google Scholar] [CrossRef]
- Cancel-Tirado, S.M.; Evans, R.B.; Yoon, K.-J. Monoclonal antibody analysis of porcine reproductive and respiratory syndrome virus epitopes associated with antibody-dependent enhancement and neutralization of virus infection. Vet. Immunol. Immunopathol. 2004, 102, 249–262. [Google Scholar] [CrossRef]
- Delputte, P.; Meerts, P.; Costers, S.; Nauwynck, H. Effect of virus-specific antibodies on attachment, internalization and infection of porcine reproductive and respiratory syndrome virus in primary macrophages. Vet. Immunol. Immunopathol. 2004, 102, 179–188. [Google Scholar] [CrossRef]
- Delputte, P.L.; Van Breedam, W.; Delrue, I.; Oetke, C.; Crocker, P.R.; Nauwynck, H.J. Porcine arterivirus attachment to the macrophage-specific receptor sialoadhesin is dependent on the sialic acid-binding activity of the N-terminal immunoglobulin domain of sialoadhesin. J. Virol. 2007, 81, 9546–9550. [Google Scholar] [CrossRef]
- Ostrowski, M.; Galeota, J.; Jar, A.; Platt, K.; Osorio, F.A.; Lopez, O. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J. Virol. 2002, 76, 4241–4250. [Google Scholar] [CrossRef]
- Plagemann, P.; Rowland, R.; Faaberg, K. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Arch. Virol. 2002, 147, 2327–2347. [Google Scholar] [CrossRef]
- Fan, B.; Liu, X.; Bai, J.; Zhang, T.; Zhang, Q.; Jiang, P. Influence of the amino acid residues at 70 in M protein of porcine reproductive and respiratory syndrome virus on viral neutralization susceptibility to the serum antibody. Virol. J. 2016, 13, 1–11. [Google Scholar] [CrossRef]
- Vanhee, M.; Costers, S.; Van Breedam, W.; Geldhof, M.F.; Van Doorsselaere, J.; Nauwynck, H.J. A variable region in GP4 of European-type porcine reproductive and respiratory syndrome virus induces neutralizing antibodies against homologous but not heterologous virus strains. Viral Immunol. 2010, 23, 403–413. [Google Scholar] [CrossRef]
- Costers, S.; Lefebvre, D.J.; Van Doorsselaere, J.; Vanhee, M.; Delputte, P.L.; Nauwynck, H.J. GP4 of porcine reproductive and respiratory syndrome virus contains a neutralizing epitope that is susceptible to immunoselection in vitro. Arch. Virol. 2010, 155, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Costers, S.; Vanhee, M.; Van Breedam, W.; Van Doorsselaere, J.; Geldhof, M.; Nauwynck, H.J. GP4-specific neutralizing antibodies might be a driving force in PRRSV evolution. Virus Res. 2010, 154, 104–113. [Google Scholar] [CrossRef]
- Wissink, E.; Kroese, M.; Van Wijk, H.; Rijsewijk, F.; Meulenberg, J.; Rottier, P. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. J. Virol. 2005, 79, 12495–12506. [Google Scholar] [CrossRef]
- Das, P.B.; Dinh, P.X.; Ansari, I.H.; De Lima, M.; Osorio, F.A.; Pattnaik, A.K. The minor envelope glycoproteins GP2a and GP4 of porcine reproductive and respiratory syndrome virus interact with the receptor CD163. J. Virol. 2010, 84, 1731–1740. [Google Scholar] [CrossRef]
- Meulenberg, J.; Van Nieuwstadt, A.; van Essen-Zandbergen, A.; Langeveld, J. Posttranslational processing and identification of a neutralization domain of the GP4 protein encoded by ORF4 of Lelystad virus. J. Virol. 1997, 71, 6061–6067. [Google Scholar] [CrossRef]
- Vashisht, K.; Goldberg, T.L.; Husmann, R.J.; Schnitzlein, W.; Zuckermann, F.A. Identification of immunodominant T-cell epitopes present in glycoprotein 5 of the North American genotype of porcine reproductive and respiratory syndrome virus. Vaccine 2008, 26, 4747–4753. [Google Scholar] [CrossRef]
- Díaz, I.; Pujols, J.; Ganges, L.; Gimeno, M.; Darwich, L.; Domingo, M.; Mateu, E. In silico prediction and ex vivo evaluation of potential T-cell epitopes in glycoproteins 4 and 5 and nucleocapsid protein of genotype-I (European) of porcine reproductive and respiratory syndrome virus. Vaccine 2009, 27, 5603–5611. [Google Scholar] [CrossRef]
- Luo, Q.; Zheng, Y.; Zhang, H.; Yang, Z.; Sha, H.; Kong, W.; Zhao, M.; Wang, N. Research progress on glycoprotein 5 of porcine reproductive and respiratory syndrome virus. Animals 2023, 13, 813. [Google Scholar] [CrossRef]
- Gong, H.-R.; Hu, Y.-f.; Li, X.; Yau, T.; Zhang, B.-Z.; Huang, J.-D. Non-neutralizing epitopes shade neutralizing epitopes against omicron in a multiple epitope-based vaccine. ACS Infect. Dis. 2022, 8, 2586–2593. [Google Scholar] [CrossRef]
- Devi, A.; Chaitanya, N.S. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J. Biomol. Struct. Dyn. 2021, 39, 6903–6917. [Google Scholar] [CrossRef]
- Ullah, H.; Ullah, S.; Li, J.; Yang, F.; Tan, L. An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes. Biology 2024, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Li, X.; Li, M.; Bi, R.; Li, Y.; Song, J.; Li, W.; Yan, M.; Luo, H.; Sun, C. In silico design of a broad-spectrum multiepitope vaccine against influenza virus. Int. J. Biol. Macromol. 2024, 254, 128071. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 2.3.1–2.3.22. [Google Scholar] [CrossRef]
- Oliver, T.; Schmidt, B.; Nathan, D.; Clemens, R.; Maskell, D. Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics 2005, 21, 3431–3432. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Middleton, D.; Menchaca, L.; Rood, H.; Komerofsky, R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003, 61, 403–407. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Gupta, S.; Vir, P.; Raghava, G.P.S. Prediction of IL4 Inducing Peptides. J. Immunol. Res. 2013, 2013, 263952. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Goboza, M.; Klein, A.; Madiehe, A.M.; Meyer, M. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci. Rep. 2021, 11, 19707. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Jalal, K.; Khan, K.; Ahmad, D.; Hayat, A.; Basharat, Z.; Abbas, M.N.; Alghamdi, S.; Almehmadi, M.; Sahibzada, M.U.K. Pan-genome reverse vaccinology approach for the design of multi-epitope vaccine construct against Escherichia albertii. Int. J. Mol. Sci. 2021, 22, 12814. [Google Scholar] [CrossRef]
- Dhanushkumar, T.; Kamaraj, B.; Vasudevan, K.; Gopikrishnan, M.; Dasegowda, K.; Rambabu, M. Structural immunoinformatics approach for rational design of a multi-epitope vaccine against triple negative breast cancer. Int. J. Biol. Macromol. 2023, 243, 125209. [Google Scholar]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T help in BALB/c mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.; Appel, R.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Wang, X. Multi-epitope vaccines: A promising strategy against viral diseases in swine. Front Cell Infect Microbiol. 2024, 14, 1497580. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham III, T.; Darden, T.; Duke, R.; Gohlke, H. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Allemailem, K.S. A Comprehensive Computer Aided Vaccine Design Approach to Propose a Multi-Epitopes Subunit Vaccine against Genus Klebsiella Using Pan-Genomics, Reverse Vaccinology, and Biophysical Techniques. Vaccines 2021, 9, 1087. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33 (Suppl. S2), W526–W531. [Google Scholar] [CrossRef]
- Liu, D.; Chen, Y. Epitope screening and vaccine molecule design of PRRSV GP3 and GP5 protein based on immunoinformatics. J. Cell Mol. Med. 2024, 28, e18103. [Google Scholar] [CrossRef]
- Cai, H.; Zhang, H.; Cheng, H.; Liu, M.; Wen, S.; Ren, J. Progress in PRRSV infection and adaptive immune response mechanisms. Viruses 2023, 15, 1442. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yi, H.; Ma, J.; Wei, Y.; Cai, M.; Li, Q.; Qin, C.; Chen, Y.-j.; Han, X.; Zhong, R. Ginsenoside Rg1 suppresses type 2 PRRSV infection via NF-κB signaling pathway in vitro, and provides partial protection against HP-PRRSV in piglet. Viruses 2019, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Wium, M.; Jonker, H.I.; Olivier, A.J.; Bellstedt, D.U.; Botes, A. DNA vaccines against Mycoplasma elicit humoral immune responses in ostriches. Front. Immunol. 2019, 10, 1061. [Google Scholar] [CrossRef] [PubMed]
- Shabir, N.; Khatun, A.; Nazki, S.; Kim, B.; Choi, E.-J.; Sun, D.; Yoon, K.-J.; Kim, W.-I. Evaluation of the cross-protective efficacy of a chimeric porcine reproductive and respiratory syndrome virus constructed based on two field strains. Viruses 2016, 8, 240. [Google Scholar] [CrossRef]
- Sira, E.M.J.S.; Banico, E.C.; Fajardo, L.E.; Odchimar, N.M.O.; Cruz, K.M.D.; Orosco, F.L. In silico design of multi-epitope vaccine candidate based on structural proteins of porcine reproductive and respiratory syndrome virus. Vet. Immunol. Immunopathol. 2025, 280, 110881. [Google Scholar] [CrossRef]
- Tseng, C.Y.; Murtada, F.; Chou, L.Y. Precision nanoscale patterning of TLR ligands for improved cancer immunotherapy. Cell Rep. Methods 2024, 4, 100782. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Protein | Start –End | Epitopes | Length | Antigenicity Score | Allergenicity /Toxicity | Intra/Inter Conservancy |
|---|---|---|---|---|---|---|---|
| PRRSV1 | ORF2A | 57–66 | APRYSVRALP | 10 | 0.8784 | Non/Non | 75.00% /20.00% |
| PRRSV2 | GP3 | 38–45 | LVRGNFSF | 8 | 1.7037 | Non/Non | 100.00% /33.33% |
| PRRSV2 | GP4 | 44–57 | QHTQQHHLVIDHIR | 14 | 0.5457 | Non/Non | 100.00% /33.33% |
| PRRSV1 | GP5 | 49–61 | LNGTDWLDKRFDW | 13 | 1.7333 | Non/Non | 75.00% /20.00% |
| PRRSV2 | M | 131–144 | RKPGLTSVNGTLV | 13 | 0.8659 | Non/Non | 100.00% /33.33% |
| PRRSV1 | N | 6–13 | HFPLAAED | 8 | 0.9417 | Non/Non | 66.00% /22.22% |
| Genotype | Protein | HLA- Allele | Epitope | Antigenicity | Allergenicity/Toxicity | IL-4/IFN- γ Induction | Intra/Inter Conservancy |
|---|---|---|---|---|---|---|---|
| PRRSV1 | ORF1a | DPA1*02:01 DPB1*05:01 | GVAPAVRIAERYRGR | 0.9596 | Non/Non | Inducer /Inducer | 100.00% /13.33% |
| PRRSV1 | ORF1a | DRB4*01:04 | KPIAYANLDEKKISA | 0.9123 | Non/Non | Inducer /Inducer | 100.00% /13.33% |
| PRRSV2 | ORF1b | DRB1*12:04 | CLGDFKQLHPVGFDS | 1.2326 | Non/Non | Inducer /Inducer | 95.24% /20.00% |
| PRRSV2 | GP2a | DPA1*02:01 DPB1*04:01 | LSFASDWFAPRYSVR | 0.5461 | Non/Non | Inducer /Inducer | 75.00% /20.00% |
| PRRSV2 | GP2b | DRB1*04:04 | VFCIRLVCSAIHRSR | 1.3347 | Non/Non | Inducer /Inducer | 100.00% /20.00% |
| PRRSV2 | GP3 | DPA1*02:01 DPB1*04:01 | NWFHLEWLRPFFSSW | 0.4583 | Non/Non | Inducer /Inducer | 75.00% /20.00% |
| PRRSV1 | GP4 | DRB5*01:03 | ACVNFTDYVAHVTQH | 1.0257 | Non/Non | Inducer /Inducer | 75.00% /13.33% |
| PRRSV2 | GP5 | DPA1*01:03 DPB1*01:01 | WRYSCTRYTNFLLDT | 0.5114 | Non/Non | Inducer /Inducer | 100.00% /13.33% |
| PRRSV1 | M | DPA1*02:01 DPB1*02:01 | LAFSITYTPIIYALK | 1.4017 | Non/Non | Inducer /Inducer | 100.00% /13.33% |
| PRRSV2 | N | DQA1*01:01 DQB1*02:01 | PHFPLATEDDVRHHF | 0.4807 | Non/Non | Inducer /Inducer | 66.67% /26.67% |
| Genotype | Protein | SLA-Allele | Epitope | Antigenicity /Immunogenicity | Allergenicity /Toxicity | Intra/Inter Conservancy |
|---|---|---|---|---|---|---|
| PRRSV1 | ORF1a | 1*1101 | AALTGRTL | 1.2071 /0.14994 | Non/Non | 100.00% /25.00% |
| PRRSV1 | ORF1a | 3*0501 | HQKPIAYANL | 1.1782 /0.1123 | Non/Non | 100.00% /20.00% |
| PRRSV2 | ORF2b | 3*0701 | TRARHAIF | 1.0977 /0.31383 | Non/Non | 95.24% /25.00% |
| PRRSV2 | GP2a | 3*0401 | SVRALPFTL | 1.5046 /0.15641 | Non/Non | 75.00% /22.22% |
| PRRSV1 | GP2b | 2*0101 | IFLAILFGF | 0.7123 /0.27353 | Non/Non | 75.00% /22.22% |
| PRRSV2 | GP3 | 2*0101 | YAWLAFLSF | 1.1215 /0.10302 | Non/Non | 100.00% /22.22% |
| PRRSV2 | GP4 | 1*0702 | SACVNFTDY | 1.6035 /0.17366 | Non/Non | 66.67% /22.22% |
| PRRSV2 | GP5 | 3*0101 | TRYTNFLL | 1.2753 /0.1324 | Non/Non | 66.67% /25.00% |
| PRRSV1 | M | 2*1201 | FSITYTPII | 1.4228 /0.1836 | Non/Non | 66.67% /22.22% |
| PRRSV1 | N | 2*1101 | FPLATEDDVRHHF | 0.4645 /0.34939 | Non/Non | 50.00% /38.46% |
| Parameters | PRRSV-V-1 Vaccine | PRRSV-V-2 Vaccine | PRRSV-V-3 Vaccine |
|---|---|---|---|
| No. of amino acids | 531 | 448 | 551 |
| Molecular weight | 57,526.93 kDa | 49,446.89 kDa | 60,733.93 kDa |
| Instability index | 22.04 | 25.72 | 28.66 |
| Aliphatic index | 81.02 | 80.91 | 77.04 |
| Half-life | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) |
| Solubility | 0.979905 | 0.975094 | 0.986037 |
| GRAVY | −0.054 | −0.191 | −0.249 |
| Theoretical pI | 9.12 | 9.82 | 9.17 |
| Antigenicity | 0.61 | 0.68 | 0.63 |
| Allergenicity | Non-allergenic | Non-allergenic | Non-allergenic |
| Toxicity | Non-toxic | Non-toxic | Non-toxic |
| Vaccine–Receptor Complex | Docking Score | Number of Residues in Interface | Area of Interface (Å2) | Number of Salt Bridges | Number of Hydrogen Bonds | Number of Non-Covalent Interactions |
|---|---|---|---|---|---|---|
| PRRSV-V-1 TLR-3 | −196.74 | 15 20 | 1104 1114 | 2 | 3 | 121 |
| PRRSV-V-1 TLR-8 | −190.43 | 10 14 | 791 870 | 0 | 2 | 87 |
| PRRSV-V-2 TLR-3 | −308.16 | 20 15 | 939 1063 | 3 | 6 | 155 |
| PRRSV-V-2 TLR-8 | −263.17 | 21 22 | 1010 975 | 1 | 9 | 176 |
| PRRSV-V-3 TLR-3 | −260.99 | 30 37 | 2129 1943 | 4 | 3 | 316 |
| PRRSV-V-3 TLR-8 | −335.61 | 34 37 | 2340 2211 | 1 | 3 | 282 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, S.; Ullah, H.; Fatima, K.; Lei, T. In Silico Designed Multi-Epitope Vaccine Based on the Conserved Fragments in Viral Proteins for Broad-Spectrum Protection Against Porcine Reproductive and Respiratory Syndrome Virus. Vet. Sci. 2025, 12, 577. https://doi.org/10.3390/vetsci12060577
Ullah S, Ullah H, Fatima K, Lei T. In Silico Designed Multi-Epitope Vaccine Based on the Conserved Fragments in Viral Proteins for Broad-Spectrum Protection Against Porcine Reproductive and Respiratory Syndrome Virus. Veterinary Sciences. 2025; 12(6):577. https://doi.org/10.3390/vetsci12060577
Chicago/Turabian StyleUllah, Shaukat, Hikmat Ullah, Kainat Fatima, and Tan Lei. 2025. "In Silico Designed Multi-Epitope Vaccine Based on the Conserved Fragments in Viral Proteins for Broad-Spectrum Protection Against Porcine Reproductive and Respiratory Syndrome Virus" Veterinary Sciences 12, no. 6: 577. https://doi.org/10.3390/vetsci12060577
APA StyleUllah, S., Ullah, H., Fatima, K., & Lei, T. (2025). In Silico Designed Multi-Epitope Vaccine Based on the Conserved Fragments in Viral Proteins for Broad-Spectrum Protection Against Porcine Reproductive and Respiratory Syndrome Virus. Veterinary Sciences, 12(6), 577. https://doi.org/10.3390/vetsci12060577