
Gut Microbiota Dynamics in Hibernating and Active Nyctalus noctula: Hibernation-Associated Loss of Diversity and Anaerobe Enrichment

 , ,
, ,  , and
, and
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. High-Throughput 16S rRNA Sequencing and Bioinformatic Data Processing
2.3. Statistical Analysis
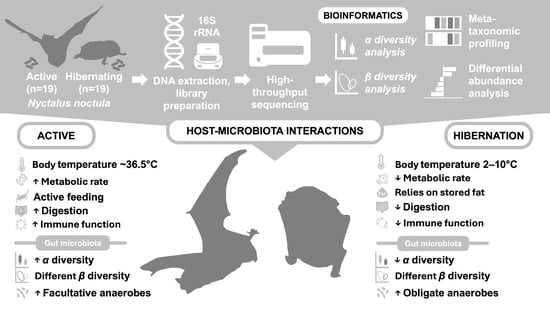
3. Results
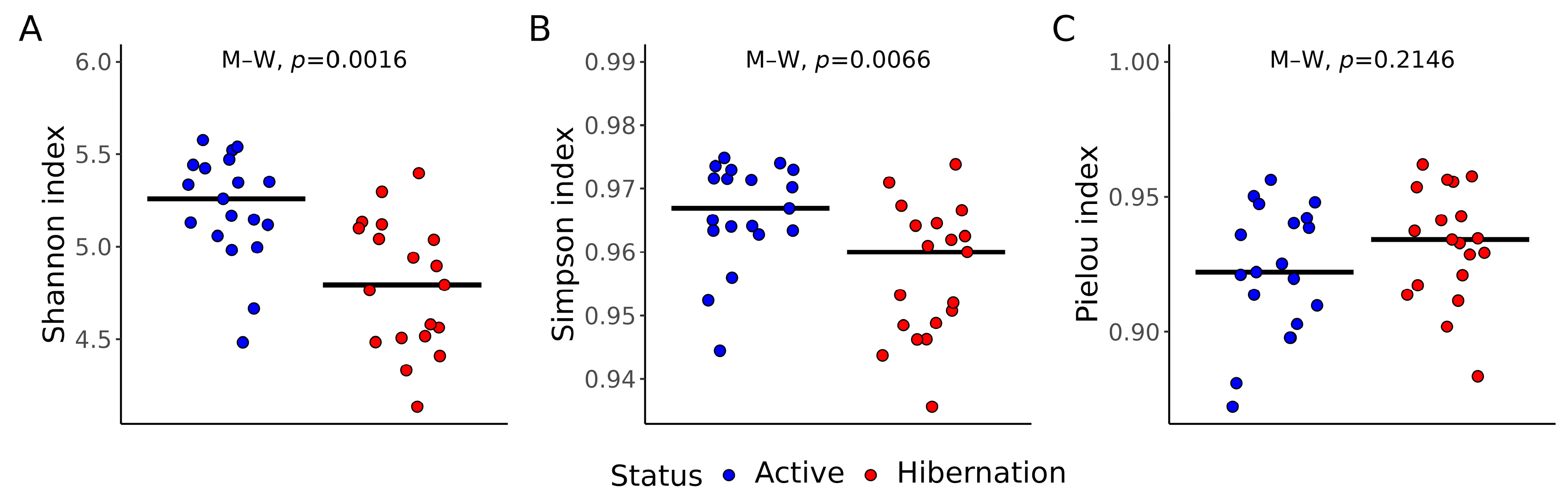
3.1. Gut Bacterial Diversity
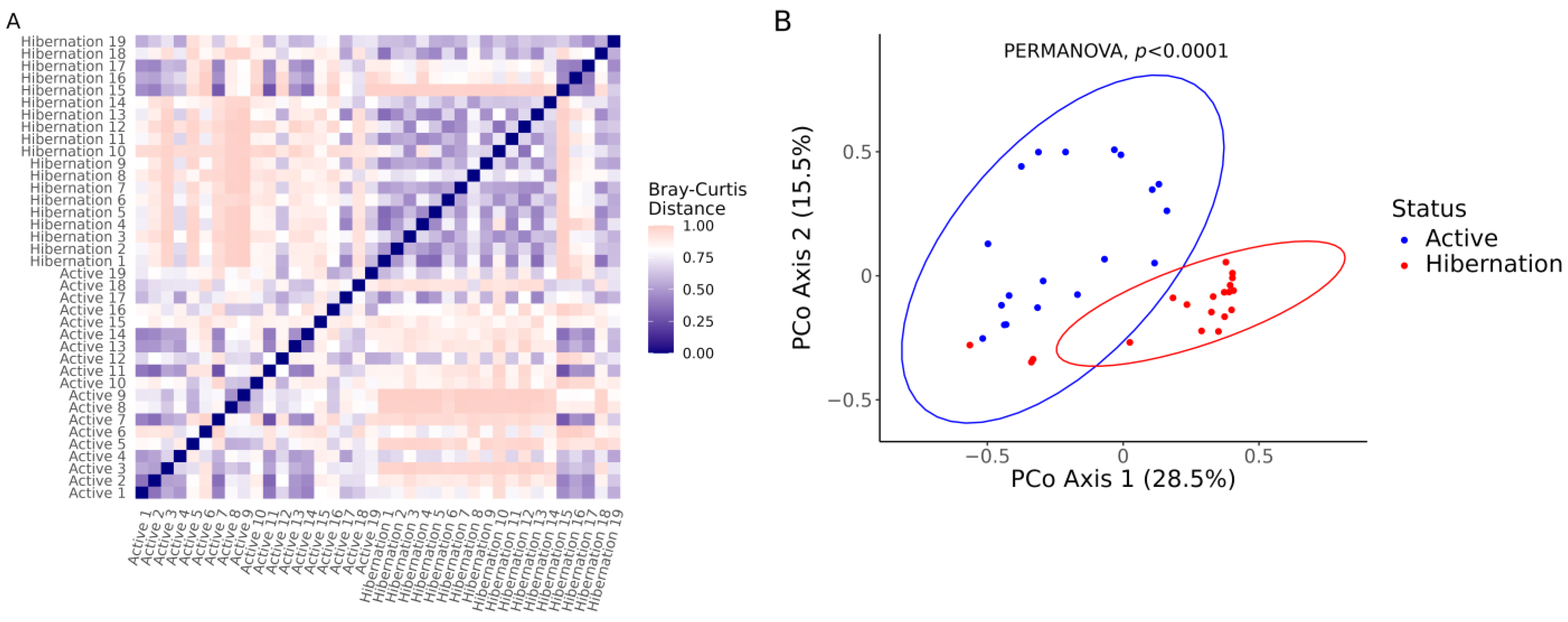
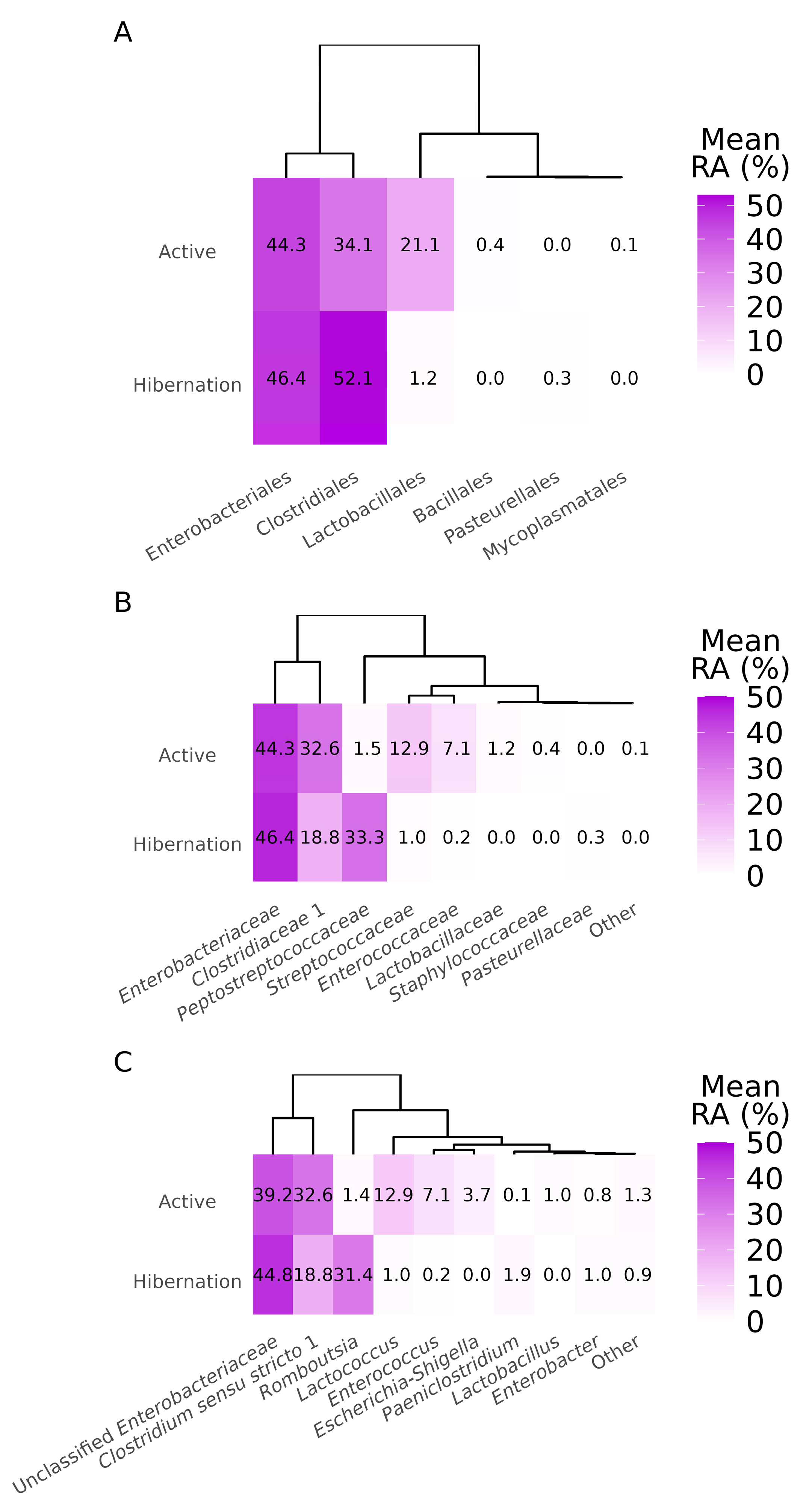
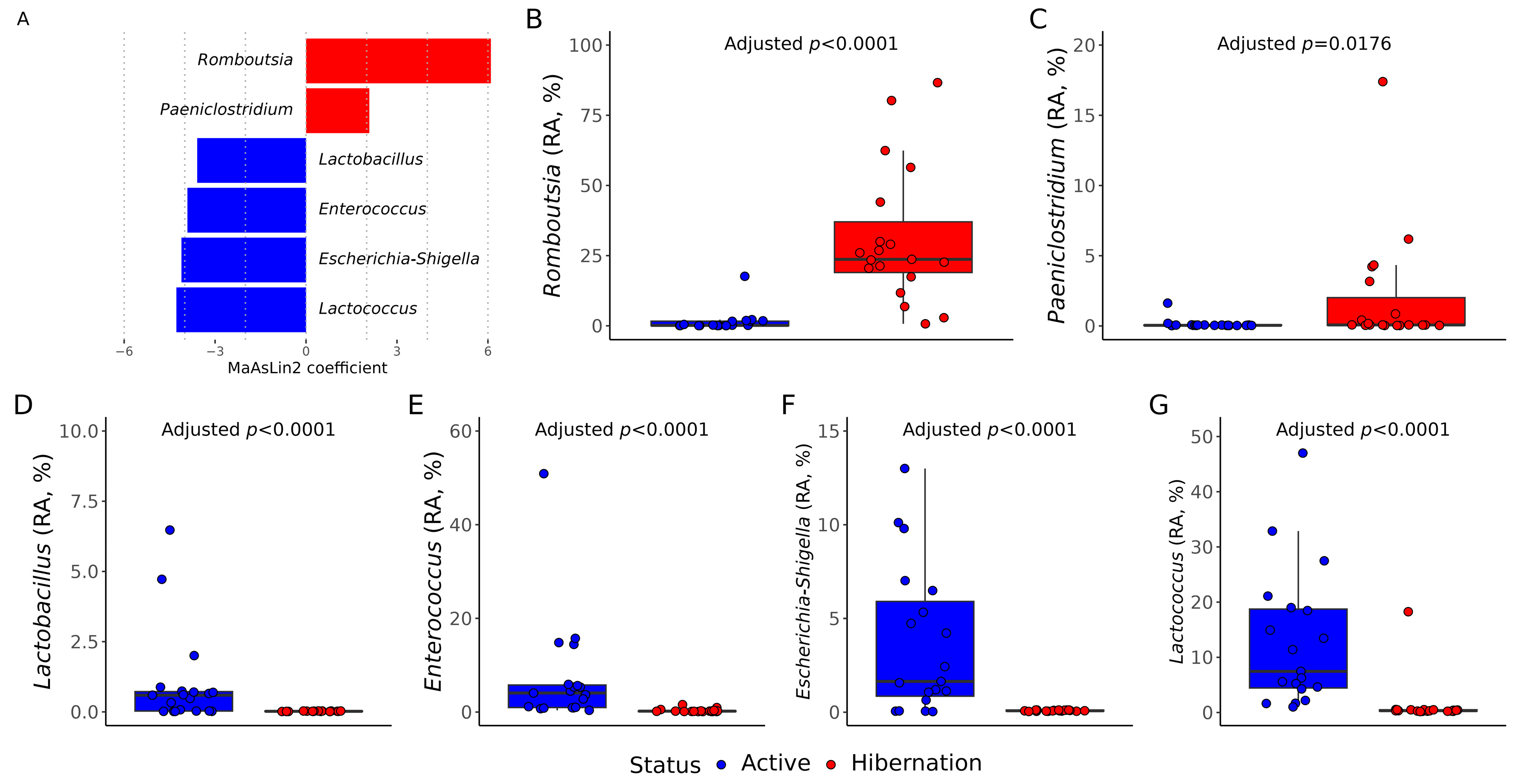
3.2. Gut Bacterial Communities Composition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S rRNA | 16S Ribosomal Ribonucleic Acid |
| DNA | Deoxyribonucleic Acid |
| RNA | Ribonucleic Acid |
| NGS | Next-Generation Sequencing |
| PCR | Polymerase Chain Reaction |
| V3-V4 | Variable Regions 3 and 4 (of the 16S rRNA gene) |
| M–W | Mann–Whitney Test |
| PERMANOVA | Permutational Multivariate Analysis of Variance |
| PCoA | Principal Coordinates Analysis |
| QIIME2 | Quantitative Insights into Microbial Ecology 2 |
| R | R Programming Language |
References
- Simmons, N.B. Order Chiroptera. Mammal Species of the World: A Taxonomic and Geographic Reference; JHU Press: Baltimore, MA, USA, 2005; Volume 1, pp. 312–529. [Google Scholar]
- Tsang, S.M.; Cirranello, A.L.; Bates, P.J.J.; Simmons, N.B. The Roles of Taxonomy and Systematics in Bat Conservation. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C.C., Kingston, T., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 503–538. ISBN 978-3-319-25220-9. [Google Scholar]
- Dumont, E.R.; Dávalos, L.M.; Goldberg, A.; Santana, S.E.; Rex, K.; Voigt, C.C. Morphological Innovation, Diversification and Invasion of a New Adaptive Zone. Proc. R. Soc. B 2012, 279, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.R.; Colocho, R.A.B.; Pereira, C.R.; Veira, T.M.; Gregorin, R.; Lage, A.P.; Dorneles, E.M.S. Zoonotic Bacterial Pathogens in Bats Samples around the World: A Scoping Review. Prev. Vet. Med. 2024, 225, 106135. [Google Scholar] [CrossRef]
- Popov, I.V.; Ohlopkova, O.V.; Donnik, I.M.; Zolotukhin, P.V.; Umanets, A.; Golovin, S.N.; Malinovkin, A.V.; Belanova, A.A.; Lipilkin, P.V.; Lipilkina, T.A.; et al. Detection of Coronaviruses in Insectivorous Bats of Fore-Caucasus, 2021. Sci. Rep. 2023, 13, 2306. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-Borne Virus Diversity, Spillover and Emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Sun, J.; Li, D.; Wang, N.; Wang, L.; Zhang, C.; Meng, X.; Ji, X.; Suchard, M.A.; Zhang, X.; et al. Emerging Viruses: Cross-Species Transmission of Coronaviruses, Filoviruses, Henipaviruses, and Rotaviruses from Bats. Cell Rep. 2022, 39, 110969. [Google Scholar] [CrossRef]
- Demian, W.L.; Cormier, O.; Mossman, K. Immunological Features of Bats: Resistance and Tolerance to Emerging Viruses. Trends Immunol. 2024, 45, 198–210. [Google Scholar] [CrossRef]
- Donnik, I.M.; Popov, I.V.; Sereda, S.V.; Popov, I.V.; Chikindas, M.L.; Ermakov, A.M. Coronavirus Infections of Animals: Future Risks to Humans. Biol. Bull. Russ. Acad. Sci. 2021, 48, 26–37. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Kennedy, B.K. The World Goes Bats: Living Longer and Tolerating Viruses. Cell Metab. 2020, 32, 31–43. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Why Do Bats Live so Long?—Possible Molecular Mechanisms. Biogerontology 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Russo, D.; Ancillotto, L. Sensitivity of Bats to Urbanization: A Review. Mamm. Biol. 2015, 80, 205–212. [Google Scholar] [CrossRef]
- Nunes, H.; Rocha, F.L.; Cordeiro-Estrela, P. Bats in Urban Areas of Brazil: Roosts, Food Resources and Parasites in Disturbed Environments. Urban Ecosyst. 2017, 20, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Dalannast, M.; Hoyt, J.R.; Byambajav, D.; Munkhtaivan, U.; Narantsetseg, N.; Batbold, B.; Jargalsaikhan, A. Human Impact and Environmental Conditions Lead to a Mass Mortality Event of David’s Myotis (Myotis Davidii) in Mongolia. Anim. Conserv. 2024, 28, 169–171. [Google Scholar] [CrossRef]
- Egert-Berg, K.; Handel, M.; Goldshtein, A.; Eitan, O.; Borissov, I.; Yovel, Y. Fruit Bats Adjust Their Foraging Strategies to Urban Environments to Diversify Their Diet. BMC Biol. 2021, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, L.M.S.; Bueno-Rocha, I.D.; Oliveira, G.; Pires, E.S.; Vasconcelos, S.; Nunes, G.L.; Frizzas, M.R.; Togni, P.H.B. Going out for Dinner—The Consumption of Agriculture Pests by Bats in Urban Areas. PLoS ONE 2021, 16, e0258066. [Google Scholar] [CrossRef]
- Stidsholt, L.; Scholz, C.; Hermanns, U.; Teige, T.; Post, M.; Stapelfeldt, B.; Reusch, C.; Voigt, C.C. Low Foraging Rates Drive Large Insectivorous Bats Away from Urban Areas. Glob. Change Biol. 2023, 30, e17063. [Google Scholar] [CrossRef]
- Dhivahar, J.; Parthasarathy, A.; Krishnan, K.; Kovi, B.S.; Pandian, G.N. Bat-Associated Microbes: Opportunities and Perils, an Overview. Heliyon 2023, 9, e22351. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Mammalian Hibernation: Biochemical Adaptation and Gene Expression. In Functional Metabolism; Storey, K.B., Ed.; Wiley: Hoboken, NJ, USA, 2004; pp. 443–471. ISBN 978-0-471-41090-4. [Google Scholar]
- Carey, H.V.; Andrews, M.T.; Martin, S.L. Mammalian Hibernation: Cellular and Molecular Responses to Depressed Metabolism and Low Temperature. Physiol. Rev. 2003, 83, 1153–1181. [Google Scholar] [CrossRef]
- Sahdo, B.; Evans, A.L.; Arnemo, J.M.; Fröbert, O.; Särndahl, E.; Blanc, S. Body Temperature during Hibernation Is Highly Correlated with a Decrease in Circulating Innate Immune Cells in the Brown Bear (Ursus Arctos): A Common Feature among Hibernators? Int. J. Med. Sci. 2013, 10, 508–514. [Google Scholar] [CrossRef]
- Bouma, H.R.; Carey, H.V.; Kroese, F.G.M. Hibernation: The Immune System at Rest? J. Leukoc. Biol. 2010, 88, 619–624. [Google Scholar] [CrossRef]
- Popov, I.V.; Berezinskaia, I.S.; Popov, I.V.; Martiusheva, I.B.; Tkacheva, E.V.; Gorobets, V.E.; Tikhmeneva, I.A.; Aleshukina, A.V.; Tverdokhlebova, T.I.; Chikindas, M.L.; et al. Cultivable Gut Microbiota in Synanthropic Bats: Shifts of Its Composition and Diversity Associated with Hibernation. Animals 2023, 13, 3658. [Google Scholar] [CrossRef]
- O’Toole, P.W.; Flemer, B. From Culture to High-Throughput Sequencing and Beyond. Gastroenterol. Clin. North Am. 2017, 46, 9–17. [Google Scholar] [CrossRef]
- Claesson, M.J.; O’Toole, P.W. Evaluating the Latest High-Throughput Molecular Techniques for the Exploration of Microbial Gut Communities. Gut Microbes 2010, 1, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.V.; Popov, I.V.; Krikunova, A.A.; Lipilkina, T.A.; Derezina, T.N.; Chikindas, M.L.; Venema, K.; Ermakov, A.M. Gut Microbiota Composition of Insectivorous Synanthropic and Fructivorous Zoo Bats: A Direct Metagenomic Comparison. Int. J. Mol. Sci. 2023, 24, 17301. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, 00191-16. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucl. Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Mann, H.B.; Whitney, D.R. On a Test of Whether One of Two Random Variables Is Stochastically Larger than the Other. Ann. Math. Statist. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA); Wiley Statsref: Statistics Reference Online; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 1–15. [Google Scholar]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package 2025. R Package Version 2.7-1. Available online: https://vegandevs.github.io/vegan/authors.html#citation (accessed on 4 June 2025).
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2; Use R!; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Carey, H.V.; Walters, W.A.; Knight, R. Seasonal Restructuring of the Ground Squirrel Gut Microbiota over the Annual Hibernation Cycle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R33–R42. [Google Scholar] [CrossRef]
- Sommer, F.; Ståhlman, M.; Ilkayeva, O.; Arnemo, J.M.; Kindberg, J.; Josefsson, J.; Newgard, C.B.; Fröbert, O.; Bäckhed, F. The Gut Microbiota Modulates Energy Metabolism in the Hibernating Brown Bear Ursus Arctos. Cell Rep. 2016, 14, 1655–1661. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, Y.; Wang, X.; Guo, D.; Wang, Y.; Wang, Y. Effects of Microhabitat Temperature Variations on the Gut Microbiotas of Free-Living Hibernating Animals. Microbiol. Spectr. 2023, 11, e00433-23. [Google Scholar] [CrossRef]
- Xiao, G.; Liu, S.; Xiao, Y.; Zhu, Y.; Zhao, H.; Li, A.; Li, Z.; Feng, J. Seasonal Changes in Gut Microbiota Diversity and Composition in the Greater Horseshoe Bat. Front. Microbiol. 2019, 10, 2247. [Google Scholar] [CrossRef]
- Gao, P.; Shen, W.; Bo, T. The Interaction between Gut Microbiota and Hibernation in Mammals. Front. Microbiol. 2024, 15, 1433675. [Google Scholar] [CrossRef]
- Gerritsen, J.; Hornung, B.; Renckens, B.; Van Hijum, S.A.F.T.; Martins Dos Santos, V.A.P.; Rijkers, G.T.; Schaap, P.J.; De Vos, W.M.; Smidt, H. Genomic and Functional Analysis of Romboutsia Ilealis CRIBT Reveals Adaptation to the Small Intestine. PeerJ 2017, 5, e3698. [Google Scholar] [CrossRef]
- Amin, A.; Modrackova, N.; Tejnecky, V.; Neuzil-Bunesova, V. Metabolic Diversity and Responses of Anteater Clostridial Isolates to Chitin-Based Substrates. Front. Vet. Sci. 2025, 12, 1459378. [Google Scholar] [CrossRef]
- Shaopeng, C.; Changze, C.; Youpeng, Q.; Baohong, M.; Meixian, Z.; Chenyue, J.; Chune, Z.; Xiangyan, W.; Jiang, H.; Bingang, S.; et al. Studies on Fatty Acids and Microbiota Characterization of the Gastrointestinal Tract of Tianzhu White Yaks. Front. Microbiol. 2025, 15, 1508468. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Stripe, L.A.; Baatz, J.E.; Wagner, C.L.; Chetta, K.E. Profiling Growth Patterns of Human Gut Microbes in Response to Preterm Human Milk and Formula. FASEB J. 2022, 36, 5571–5589. [Google Scholar] [CrossRef]
- Stege, P.B.; Hordijk, J.; Sandholt, A.K.S.; Zomer, A.L.; Viveen, M.C.; Rogers, M.R.C.; Salomons, M.; Wagenaar, J.A.; Mughini-Gras, L.; Willems, R.J.L.; et al. Gut Colonization by ESBL-Producing Escherichia Coli in Dogs Is Associated with a Distinct Microbiome and Resistome Composition. Microbiol. Spectr. 2023, 11, e0006323. [Google Scholar] [CrossRef]
- Zoghi, A.; Massoud, R.; Todorov, S.D.; Chikindas, M.L.; Popov, I.; Smith, S.; Khosravi-Darani, K. Role of the Lactobacilli in Food Bio-Decontamination: Friends with Benefits. Enzym. Microb. Technol. 2021, 150, 109861. [Google Scholar] [CrossRef]
- Tadesse, B.T.; Svetlicic, E.; Zhao, S.; Berhane, N.; Jers, C.; Solem, C.; Mijakovic, I. Bad to the Bone?—Genomic Analysis of Enterococcus Isolates from Diverse Environments Reveals That Most Are Safe and Display Potential as Food Fermentation Microorganisms. Microbiol. Res. 2024, 283, 127702. [Google Scholar] [CrossRef] [PubMed]
- Todorov, S.D.; Popov, I.; Weeks, R.; Chikindas, M.L. Use of Bacteriocins and Bacteriocinogenic Beneficial Organisms in Food Products: Benefits, Challenges, Concerns. Foods 2022, 11, 3145. [Google Scholar] [CrossRef]
- Vieira, G.; Sabarly, V.; Bourguignon, P.-Y.; Durot, M.; Le Fèvre, F.; Mornico, D.; Vallenet, D.; Bouvet, O.; Denamur, E.; Schachter, V.; et al. Core and Panmetabolism in Escherichia Coli. J. Bacteriol. 2011, 193, 1461–1472. [Google Scholar] [CrossRef]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The Population Genetics of Pathogenic Escherichia Coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef]
- Popov, I.V.; Mazanko, M.S.; Kulaeva, E.D.; Golovin, S.N.; Malinovkin, A.V.; Aleshukina, I.S.; Aleshukina, A.V.; Prazdnova, E.V.; Tverdokhlebova, T.I.; Chikindas, M.L.; et al. Gut Microbiota of Bats: Pro-Mutagenic Properties and Possible Frontiers in Preventing Emerging Disease. Sci. Rep. 2021, 11, 21075. [Google Scholar] [CrossRef]
- Greay, T.L.; Gofton, A.W.; Zahedi, A.; Paparini, A.; Linge, K.L.; Joll, C.A.; Ryan, U.M. Evaluation of 16S Next-Generation Sequencing of Hypervariable Region 4 in Wastewater Samples: An Unsuitable Approach for Bacterial Enteric Pathogen Identification. Sci. Total Environ. 2019, 670, 1111–1124. [Google Scholar] [CrossRef]
- Lauritsen, J.G.; Hansen, M.L.; Bech, P.K.; Jelsbak, L.; Gram, L.; Strube, M.L. Identification and Differentiation of Pseudomonas Species in Field Samples Using an rpoD Amplicon Sequencing Methodology. mSystems 2021, 6, e0070421. [Google Scholar] [CrossRef] [PubMed]
- Commichaux, S.; Luan, T.; Muralidharan, H.S.; Pop, M. Database Size Positively Correlates with the Loss of Species-Level Taxonomic Resolution for the 16S rRNA and Other Prokaryotic Marker Genes. PLoS Comput. Biol. 2024, 20, e1012343. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Teige, T.; Ngoufack Djoumessi, K.P.; Buchholz, S.; Pritsch, F.; Planillo, A.; Voigt, C.C. Dietary Diversification of an Insect Predator along an Urban-Rural Gradient. Landsc. Urban. Plan. 2025, 256, 105273. [Google Scholar] [CrossRef]
- Popov, I.V.; Popov, I.V.; Chebotareva, I.P.; Tikhmeneva, I.A.; Peshkova, D.A.; Krikunova, A.A.; Tkacheva, E.V.; Algburi, A.R.; Abdulhameed, A.M.; Jargalsaikhan, A.; et al. Differences in Gut Microbiota Composition, Diversity, and Predicted Functional Activity between Wild and Captive Zoo Carollia Perspicillata in a One Health Perspective. Braz. J. Microbiol. 2025, 56, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xiao, G.; Liu, H.; Zhao, X.; Sun, C.; Tan, X.; Sun, K.; Liu, S.; Feng, J. Captivity Causes Taxonomic and Functional Convergence of Gut Microbial Communities in Bats. PeerJ 2019, 7, e6844. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, I.V.; Peshkova, D.A.; Lukbanova, E.A.; Tsurkova, I.S.; Emelyantsev, S.A.; Krikunova, A.A.; Malinovkin, A.V.; Chikindas, M.L.; Ermakov, A.M.; Popov, I.V. Gut Microbiota Dynamics in Hibernating and Active Nyctalus noctula: Hibernation-Associated Loss of Diversity and Anaerobe Enrichment. Vet. Sci. 2025, 12, 559. https://doi.org/10.3390/vetsci12060559
Popov IV, Peshkova DA, Lukbanova EA, Tsurkova IS, Emelyantsev SA, Krikunova AA, Malinovkin AV, Chikindas ML, Ermakov AM, Popov IV. Gut Microbiota Dynamics in Hibernating and Active Nyctalus noctula: Hibernation-Associated Loss of Diversity and Anaerobe Enrichment. Veterinary Sciences. 2025; 12(6):559. https://doi.org/10.3390/vetsci12060559
Chicago/Turabian StylePopov, Ilia V., Daria A. Peshkova, Ekaterina A. Lukbanova, Inna S. Tsurkova, Sergey A. Emelyantsev, Anastasya A. Krikunova, Aleksey V. Malinovkin, Michael L. Chikindas, Alexey M. Ermakov, and Igor V. Popov. 2025. "Gut Microbiota Dynamics in Hibernating and Active Nyctalus noctula: Hibernation-Associated Loss of Diversity and Anaerobe Enrichment" Veterinary Sciences 12, no. 6: 559. https://doi.org/10.3390/vetsci12060559
APA StylePopov, I. V., Peshkova, D. A., Lukbanova, E. A., Tsurkova, I. S., Emelyantsev, S. A., Krikunova, A. A., Malinovkin, A. V., Chikindas, M. L., Ermakov, A. M., & Popov, I. V. (2025). Gut Microbiota Dynamics in Hibernating and Active Nyctalus noctula: Hibernation-Associated Loss of Diversity and Anaerobe Enrichment. Veterinary Sciences, 12(6), 559. https://doi.org/10.3390/vetsci12060559