
Isolation and Characterization of Porcine Epidemic Diarrhea Virus G2c Strains Circulating in China from 2021 to 2024

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Viral RNA Isolation and RT-qPCR Detection
2.3. PEDV Isolation
2.4. Immunofluorescence Assay
2.5. Virus TCID50 Assay and Serum Neutralization Test
2.6. Sequencing, Multiple-Sequence Alignment, and Phylogenetic Analysis
2.7. Amino Acid Sequence Analysis of the Spike Protein
2.8. Viral Recombination Analysis
3. Results
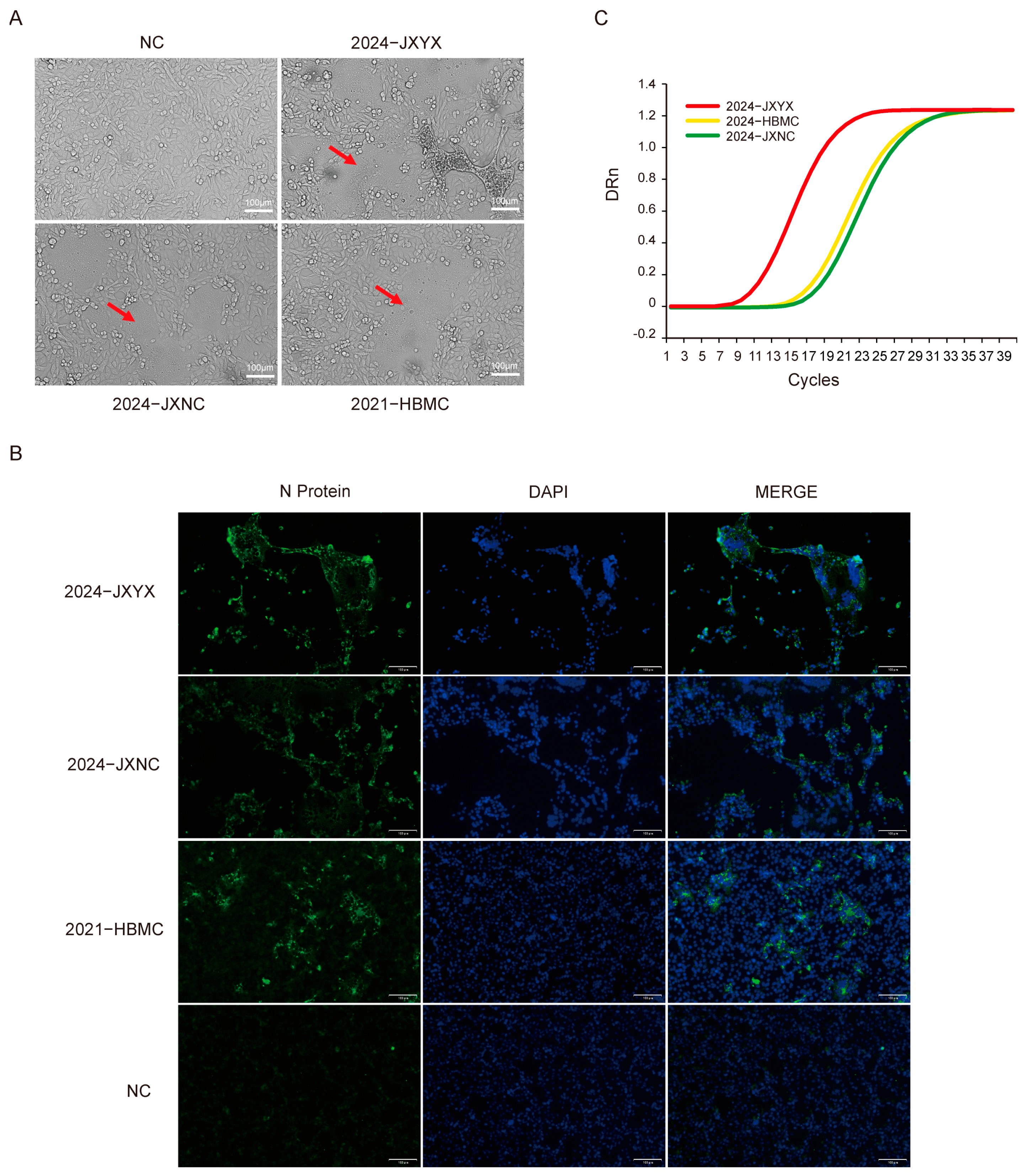
3.1. Viral Isolation and Identification
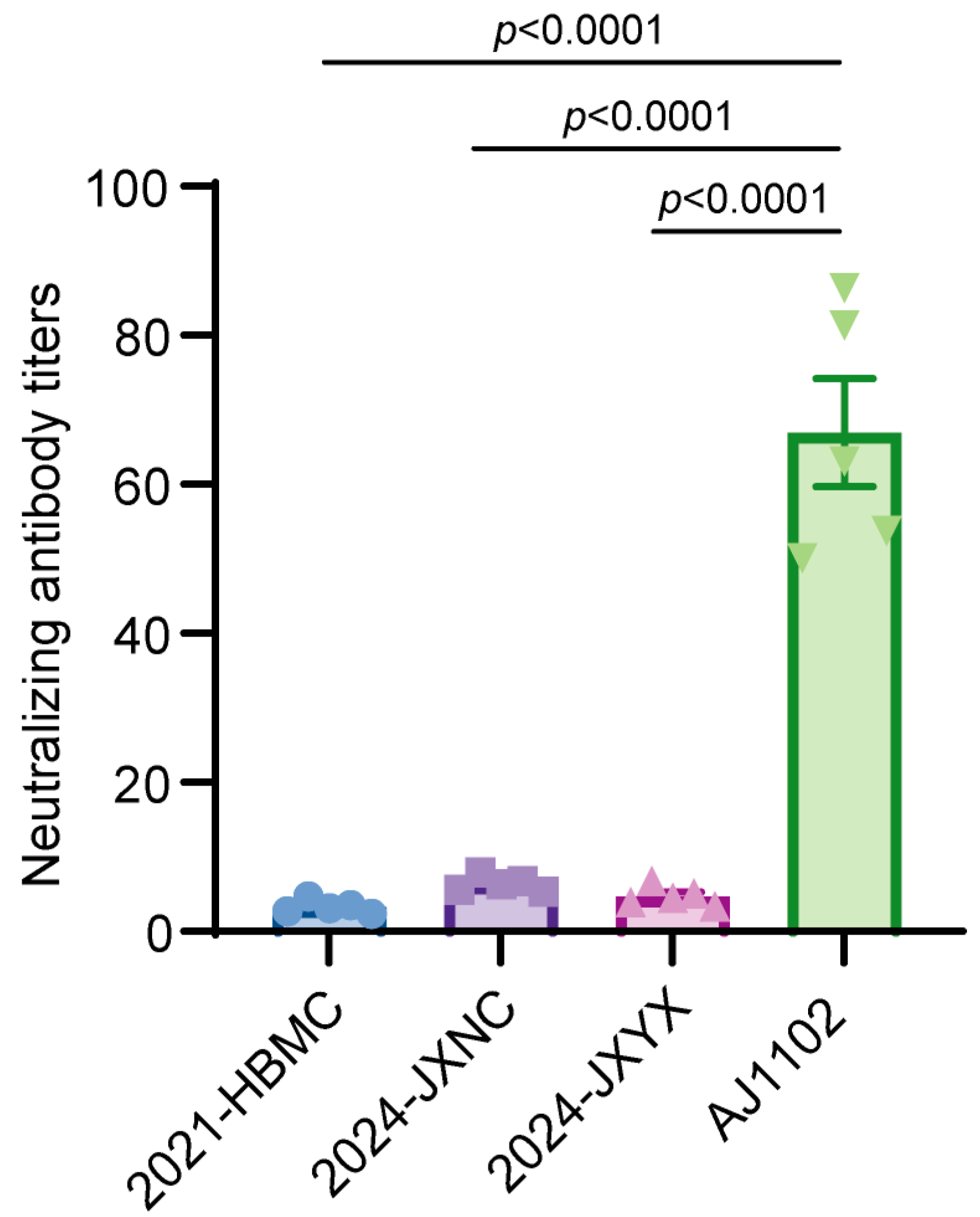
3.2. Neutralization Titer of AJ1102 Immune Serum Against Isolated Strains Is Significantly Reduced
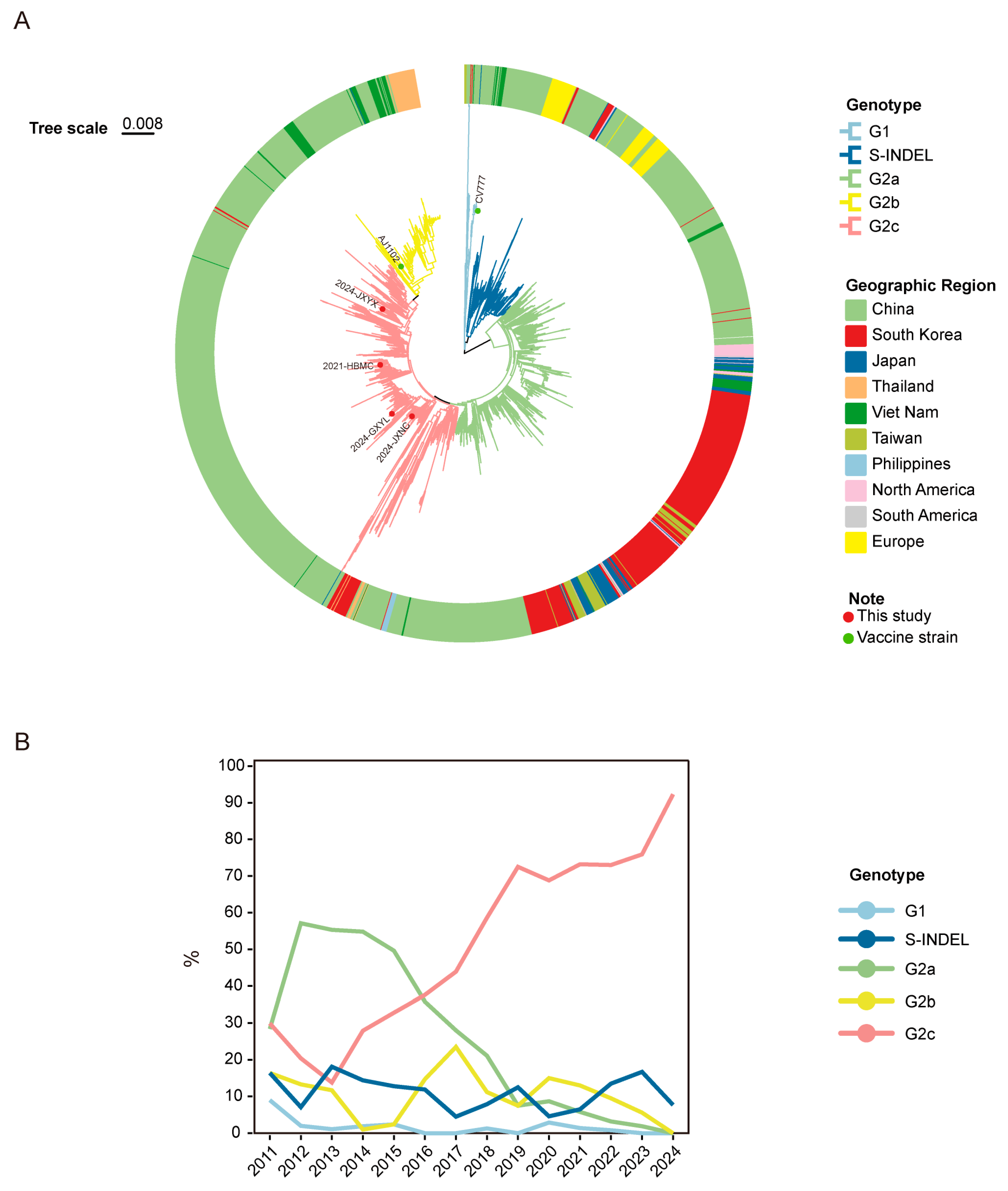
3.3. Identity and Phylogenetic Analysis of the S Gene
3.4. Comparative Analysis of Amino Acid Sequences of the Spike Protein
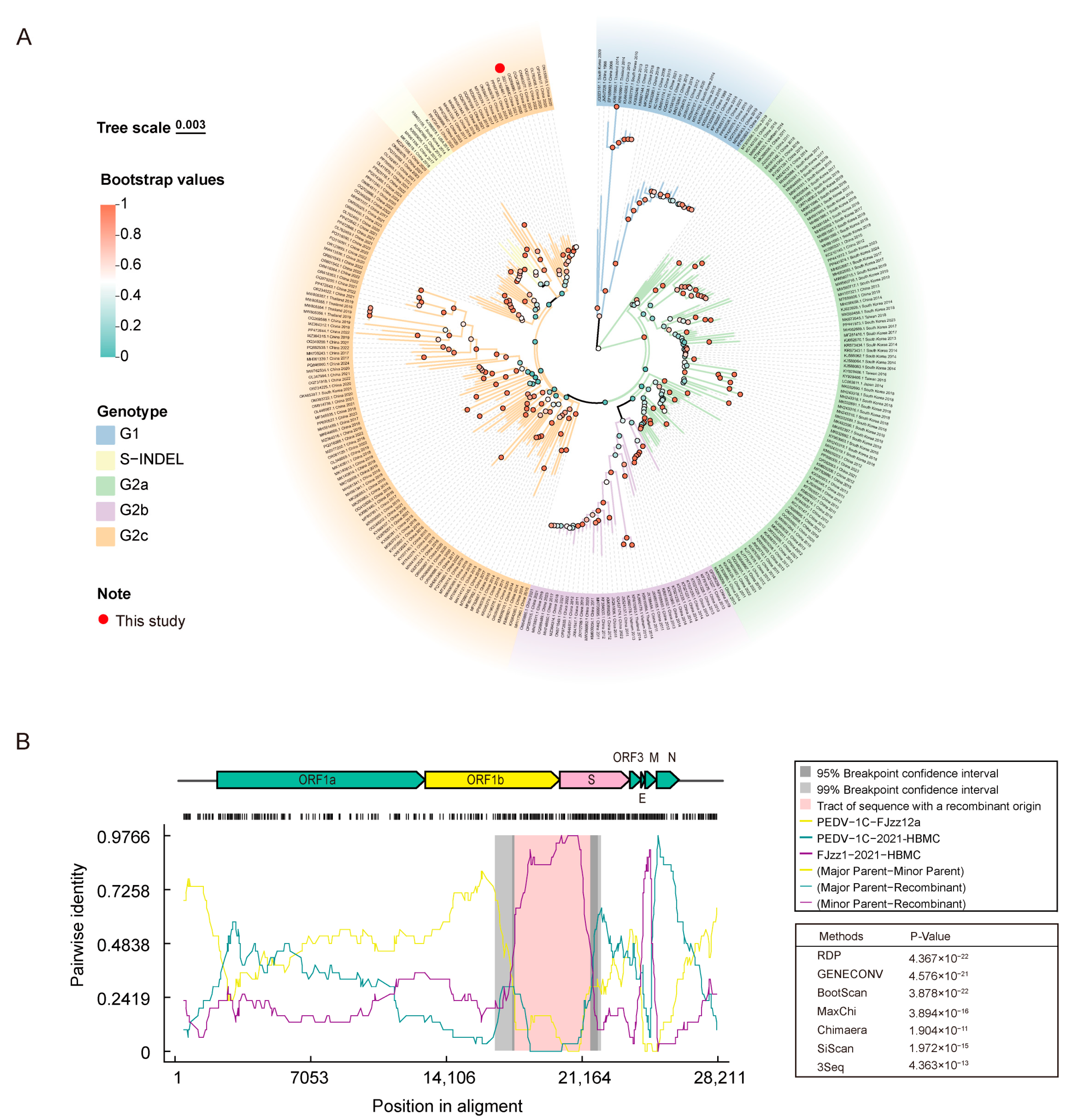
3.5. Comparative Analysis of the Complete Genome of the 2021-HBMC Strain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, S.; Liu, G.; Savelkoul, H.F.J.; Jansen, C.A.; Li, B. Mini-review: Microbiota have potential to prevent PEDV infection by improved intestinal barrier. Front. Immunol. 2023, 14, 1230937. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef]
- Li, Q.; Xu, Z.; Wu, T.; Peng, O.; Huang, L.; Zhang, Y.; Xue, C.; Wen, Z.; Zhou, Q.; Cao, Y. A flagellin-adjuvanted PED subunit vaccine improved protective efficiency against PEDV variant challenge in pigs. Vaccine 2018, 36, 4228–4235. [Google Scholar] [CrossRef]
- Shibata, I.; Tsuda, T.; Mori, M.; Ono, M.; Sueyoshi, M.; Uruno, K. Isolation of porcine epidemic diarrhea virus in porcine cell cultures and experimental infection of pigs of different ages. Vet. Microbiol. 2000, 72, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Song, D.; Zhou, X.; Peng, Q.; Chen, Y.; Zhang, F.; Huang, T.; Zhang, T.; Li, A.; Huang, D.; Wu, Q.; et al. Newly Emerged Porcine Deltacoronavirus Associated With Diarrhoea in Swine in China: Identification, Prevalence and Full-Length Genome Sequence Analysis. Transbound. Emerg. Dis. 2015, 62, 575–580. [Google Scholar] [CrossRef]
- Lin, C.M.; Saif, L.J.; Marthaler, D.; Wang, Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016, 226, 20–39. [Google Scholar] [CrossRef] [PubMed]
- He, W.-T.; Bollen, N.; Xu, Y.; Zhao, J.; Dellicour, S.; Yan, Z.; Gong, W.; Zhang, C.; Zhang, L.; Lu, M.; et al. Phylogeography Reveals Association between Swine Trade and the Spread of Porcine Epidemic Diarrhea Virus in China and across the World. Mol. Biol. Evol. 2022, 39, 364. [Google Scholar] [CrossRef]
- Kocherhans, R.; Bridgen, A.; Ackermann, M.; Tobler, K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 2001, 23, 137–144. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.-J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef]
- Wood, E.N. An apparently new syndrome of porcine epidemic diarrhoea. Vet. Rec. 1977, 100, 243–244. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Xiao, S. Porcine epidemic diarrhea in China. Virus Res. 2016, 226, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E. Porcine Epidemic Diarrhea: Insights and Progress on Vaccines. Vaccines 2024, 12, 212. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Shi, D.; Shi, H.; Zhang, X.; Li, C.; Chi, Y.; Feng, L. Detection and molecular diversity of spike gene of porcine epidemic diarrhea virus in China. Viruses 2013, 5, 2601–2613. [Google Scholar] [CrossRef]
- Guo, J.; Fang, L.; Ye, X.; Chen, J.; Xu, S.; Zhu, X.; Miao, Y.; Wang, D.; Xiao, S. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound. Emerg. Dis. 2019, 66, 111–118. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef]
- Wang, L.; Byrum, B.; Zhang, Y. New variant of porcine epidemic diarrhea virus, United States, 2014. Emerg. Infect. Dis. 2014, 20, 917–919. [Google Scholar] [CrossRef]
- Li, W.; van Kuppeveld, F.J.M.; He, Q.; Rottier, P.J.M.; Bosch, B.J. Cellular entry of the porcine epidemic diarrhea virus. Virus Res. 2016, 226, 117–127. [Google Scholar] [CrossRef]
- Hou, Y.; Lin, C.-M.; Yokoyama, M.; Yount, B.L.; Marthaler, D.; Douglas, A.L.; Ghimire, S.; Qin, Y.; Baric, R.S.; Saif, L.J.; et al. Deletion of a 197-Amino-Acid Region in the N-Terminal Domain of Spike Protein Attenuates Porcine Epidemic Diarrhea Virus in Piglets. J. Virol. 2017, 91, 227. [Google Scholar] [CrossRef]
- Li, Z.; Ma, Z.; Dong, L.; Yang, T.; Li, Y.; Jiao, D.; Han, W.; Zheng, H.; Xiao, S. Molecular Mechanism of Porcine Epidemic Diarrhea Virus Cell Tropism. mBio 2022, 13, e0373921. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Li, J.; Zhang, Y.; Zhou, Q.; Gong, L.; Xue, C.; Cao, Y. Genetic epidemiology of porcine epidemic diarrhoea virus circulating in China in 2012–2017 based on spike gene. Transbound. Emerg. Dis. 2018, 65, 883–889. [Google Scholar] [CrossRef]
- Su, M.; Li, C.; Qi, S.; Yang, D.; Jiang, N.; Yin, B.; Guo, D.; Kong, F.; Yuan, D.; Feng, L.; et al. A molecular epidemiological investigation of PEDV in China: Characterization of co-infection and genetic diversity of S1-based genes. Transbound. Emerg. Dis. 2020, 67, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Zeng, S.; Xiao, S.; Chen, H.; Fang, L. Complete genome sequence of porcine epidemic diarrhea virus strain AJ1102 isolated from a suckling piglet with acute diarrhea in China. J. Virol. 2012, 86, 10910–10911. [Google Scholar] [CrossRef]
- Won, H.; Lim, J.; Noh, Y.H.; Yoon, I.; Yoo, H.S. Efficacy of Porcine Epidemic Diarrhea Vaccines: A Systematic Review and Meta-Analysis. Vaccines 2020, 8, 642. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Huang, J.; Yao, Y.; Zhao, W.; Zhang, Y.; Qing, J.; Ren, J.; Yan, Z.; Wang, Z.; et al. Isolation and oral immunogenicity assessment of porcine epidemic diarrhea virus NH-TA2020 strain: One of the predominant strains circulating in China from 2017 to 2021. Virol. Sin. 2022, 37, 646–655. [Google Scholar] [CrossRef]
- Yang, C.; Sun, J.-Y.; Li, X.L.; Cheng, N.; Wang, K.-Y.; Li, L.-Q.; Cheng, X.-J.; Sun, Y.-F. Emerging and re-emerging genotype 2c porcine epidemic diarrhoea virus with high pathogenicity in China. J. Infect. 2024, 89, 106192. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gong, T.; Wu, D.; Feng, Y.; Gao, Q.; Xing, J.; Zheng, X.; Song, Z.; Liu, X.; Chen, X.; et al. Isolation, identification, and pathogenicity of porcine epidemic diarrhea virus. Front. Microbiol. 2023, 14, 1273589. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Yu, J.; Chai, X.; Cheng, Y.; Xing, G.; Liao, A.; Du, L.; Wang, Y.; Lei, J.; Gu, J.; Zhou, J. Molecular characteristics of the spike gene of porcine epidemic diarrhoea virus strains in Eastern China in 2016. Virus Res. 2018, 247, 47–54. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, X.; Li, H.; Ma, B.; Guan, R.; Yang, J.; Chen, D.; Han, X.; Zhou, L.; Song, Z.; et al. Molecular characterization of porcine epidemic diarrhea virus associated with outbreaks in southwest China during 2014–2018. Transbound. Emerg. Dis. 2021, 68, 3482–3497. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Fu, P.; Zhou, Y.; Lang, Y.; Zhao, S.; Wen, Y.; Wang, Y.; Wu, R.; Zhao, Q.; Du, S.; et al. Phylogenetic Analysis of Porcine Epidemic Diarrhea Virus (PEDV) during 2020-2022 and Isolation of a Variant Recombinant PEDV Strain. Int. J. Mol. Sci. 2024, 25, 10878. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Yan, W.; Yang, X.; Fan, H. Molecular characterization of porcine epidemic diarrhea virus in Sichuan from 2023 to 2024. Microb. Pathog. 2025, 203, 107486. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Zhou, D.; Geng, C.; Yang, K.; Duan, Z.; Guo, R.; Liu, W.; Yuan, F.; Liu, Z.; Gao, T.; et al. Isolation and evolutionary analyses of porcine epidemic diarrhea virus in Asia. PeerJ 2020, 8, e10114. [Google Scholar] [CrossRef]
- Su, M.; Wang, Y.; Yan, J.; Xu, X.; Zheng, H.; Cheng, J.; Du, X.; Liu, Y.; Ying, J.; Zhao, Y.; et al. Isolation and characterization of a novel S1-gene insertion porcine epidemic diarrhea virus with low pathogenicity in newborn piglets. Virulence 2024, 15, 2397512. [Google Scholar] [CrossRef]
- Sun, J.; Cheng, J.; Shi, D.; Xu, X.; Liu, Y.; Ying, J.; Zhao, Y.; Zheng, H.; Yan, J.; Sun, D.; et al. Genetic Epidemiology of Porcine Epidemic Diarrhea Virus Circulating in China From 2010 to 2024, Characterization of Phylogenetic and Genetic Diversity of S1-Based Genes. J. Med. Virol. 2025, 97, e70198. [Google Scholar] [CrossRef]
- Le, B.T.; Gallage, H.C.; Kim, M.-H.; Park, J.-E. Molecular Characterization of Porcine Epidemic Diarrhea Virus from Field Samples in South Korea. Viruses 2023, 15, 2428. [Google Scholar] [CrossRef]
- Zhu, H.; Lou, J.; Yang, Z.; Bai, J.; Jiang, P.; Wang, X.; Liu, X. STT3B promotes porcine epidemic diarrhea virus replication by regulating N-glycosylation of PEDV S protein. J. Virol. 2025, 99, e0001825. [Google Scholar] [CrossRef]
- Huang, H.-C.; Lai, Y.-J.; Liao, C.-C.; Wang, F.-Y.; Huang, K.-B.; Lee, I.-J.; Chou, W.-C.; Wang, S.-H.; Wang, L.-H.; Hsu, J.-M.; et al. Targeting conserved N-glycosylation blocks SARS-CoV-2 variant infection in vitro. EBioMedicine 2021, 74, 103712. [Google Scholar] [CrossRef]
- Aloor, A.; Aradhya, R.; Venugopal, P.; Gopalakrishnan Nair, B.; Suravajhala, R. Glycosylation in SARS-CoV-2 variants: A path to infection and recovery. Biochem. Pharmacol. 2022, 206, 115335. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, D.; Shi, W.; Lu, R.; Wang, W.; Zhao, Y.; Deng, Y.; Zhou, W.; Ren, H.; Wu, J.; et al. Origin and Possible Genetic Recombination of the Middle East Respiratory Syndrome Coronavirus from the First Imported Case in China: Phylogenetics and Coalescence Analysis. mBio 2015, 6, e01280-15. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yin, X.; Tian, H.; Qiu, Y.; Wang, Z.; Chen, J.; Ma, D.; Zhao, B.; Du, Q.; Tong, D.; et al. The S protein of a novel recombinant PEDV strain promotes the infectivity and pathogenicity of PEDV in mid-west China. Transbound. Emerg. Dis. 2022, 69, 3704–3723. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEDV Strains | CV777 | AJ1102 |
|---|---|---|
| 2021-HBMC | 93.1% | 97.2% |
| 2024-GXYL | 92.1% | 96.0% |
| 2024-JXNC | 93.2% | 97.2% |
| 2024-JXYX | 93.0% | 97.4% |
| Genotype | Strains | High-Specificity N-Glycosylation Sites | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 57 | 62 | 112 | 118 | 127 | 212 | 300 | 320 | 347 | 380 | 510 | 552 | 777 | 1245 | 1258 | ||
| G1b | KT323979-CV777 | – | – | NTSA | – | NKTL | NVTS | – | – | NSSD | – | NITV | NVTN | NISI | NKTL | – |
| G1b | KC109141-JS2008 | – | – | NTSA | – | NKTL | NVTS | – | – | NSSD | – | NITV | NVTN | NISI | NKTL | – |
| G1a | JQ023161-DR13 | – | – | – | – | NKTL | NVTS | – | NDTS | NSSD | – | NITV | NVTN | NISI | NKTL | NRTG |
| S-INDEL | KU847996-ZL29 | – | – | – | – | NKTL | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| S-INDEL | KJ399978-OH851 | – | – | – | – | NKTL | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| S-INDEL | MH243316-KNU-1702 | NSSS | – | – | – | NKTL | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| G2b | JX188454-AJ1102 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSD | – | NITV | – | NISI | NKTL | NRTG |
| G2b | KU646831-AH2012_12 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSD | – | NITV | – | NISI | NKTL | NRTG |
| G2a | JN825712-BJ-2011-1 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| G2a | KC210145-AH2012 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSN | – | – | – | NISI | NKTL | NRTG |
| G2a | MH061342-CH_SCZJ_2018 | – | NSTW | – | NATA | – | NVTS | NKTI | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| G2c | KU252649-YC2014 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| G2c | MH161337-CH_SCZG_2017 | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSN | NSTV | – | – | NISI | NKTL | NRTG |
| G2c | 2021-HBMC | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSD | – | – | – | NISI | NKTL | NRTG |
| G2c | 2024-GXYL | – | NSTW | – | NATA | – | NVTS | – | NDTS | – | – | – | – | NISI | NKTL | NRTV |
| G2c | 2024-JXNC | – | NSTW | – | NATA | – | NVTS | – | NDTS | NSSN | – | – | – | NISI | NKTL | NRTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Chen, C.; Wang, Z.; Zhang, A. Isolation and Characterization of Porcine Epidemic Diarrhea Virus G2c Strains Circulating in China from 2021 to 2024. Vet. Sci. 2025, 12, 444. https://doi.org/10.3390/vetsci12050444
Lu X, Chen C, Wang Z, Zhang A. Isolation and Characterization of Porcine Epidemic Diarrhea Virus G2c Strains Circulating in China from 2021 to 2024. Veterinary Sciences. 2025; 12(5):444. https://doi.org/10.3390/vetsci12050444
Chicago/Turabian StyleLu, Xi, Chen Chen, Zixuan Wang, and Anding Zhang. 2025. "Isolation and Characterization of Porcine Epidemic Diarrhea Virus G2c Strains Circulating in China from 2021 to 2024" Veterinary Sciences 12, no. 5: 444. https://doi.org/10.3390/vetsci12050444
APA StyleLu, X., Chen, C., Wang, Z., & Zhang, A. (2025). Isolation and Characterization of Porcine Epidemic Diarrhea Virus G2c Strains Circulating in China from 2021 to 2024. Veterinary Sciences, 12(5), 444. https://doi.org/10.3390/vetsci12050444