
Exploring the Most Effective Strategy for Purine Metabolite Quantification in Veterinary Medicine Using LC–MS/MS


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Standard and Working Solutions
2.3. Animal Samples
2.4. Sample Preparation
2.4.1. Serum Samples (Canine)
2.4.2. Urine Samples (Canine and Bovine)
2.5. LC–MS/MS Analysis
2.6. Method Validation
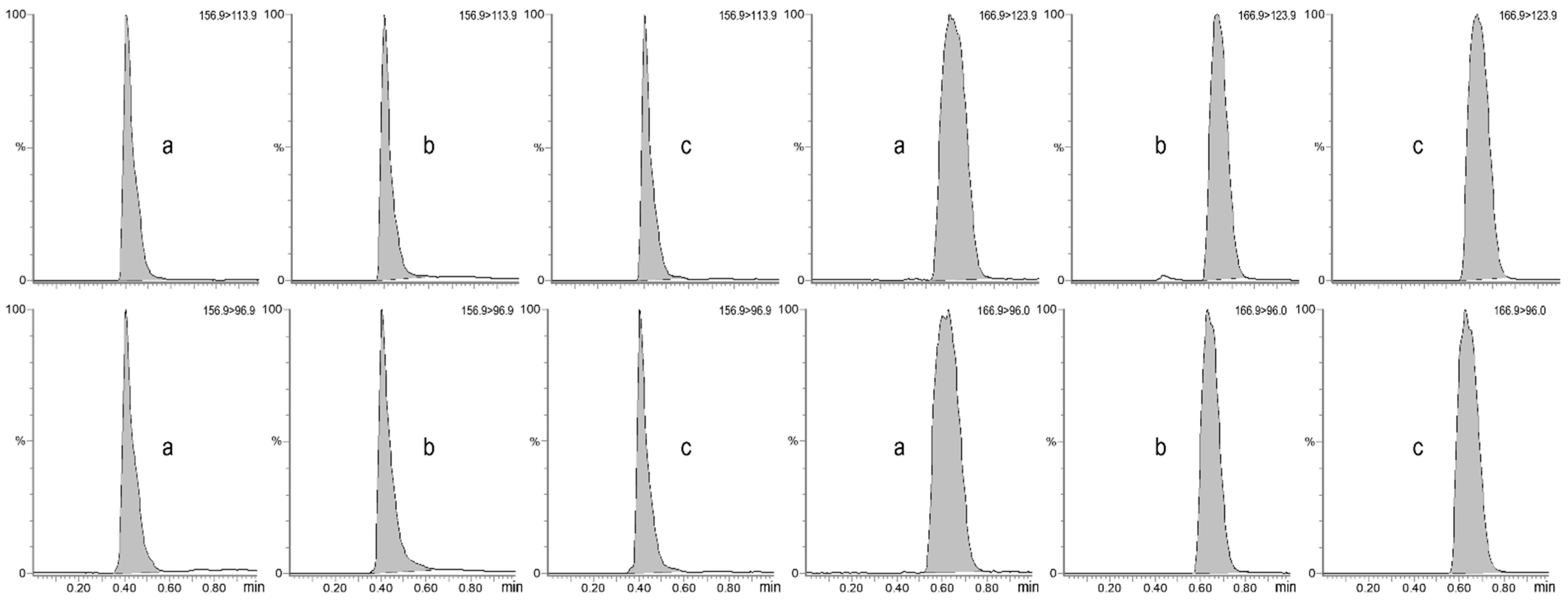
2.6.1. Specificity and Selectivity
2.6.2. Calibration Range
2.6.3. Lower Limit of Quantification (LLOQ)
2.6.4. Accuracy and Precision
2.6.5. Matrix Effect (ME)
2.6.6. Recovery
2.6.7. Stability
2.6.8. Carry-Over
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ngo, T.C.; Assimos, D.G. Uric Acid Nephrolithiasis: Recent Progress and Future Directions. Rev. Urol. 2007, 9, 17–27. [Google Scholar] [PubMed]
- Chen, C.; Lü, J.-M.; Yao, Q. Hyperuricemia-Related Diseases and Xanthine Oxidoreductase (XOR) Inhibitors: An Overview. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2501–2512. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Micheli, V.; Ipata, P.L.; Tozzi, M.G. Pediatric Neurological Syndromes and Inborn Errors of Purine Metabolism. Neurochem. Int. 2010, 56, 367–378. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. The Role of Xanthine Oxidoreductase and Uric Acid in Metabolic Syndrome. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 2557–2565. [Google Scholar] [CrossRef]
- Karmi, N.; Safra, N.; Young, A.; Bannasch, D.L. Validation of a Urine Test and Characterization of the Putative Genetic Mutation for Hyperuricosuria in Bulldogs and Black Russian Terriers. Am. J. Vet. Res. 2010, 71, 909–914. [Google Scholar] [CrossRef]
- Bannasch, D.; Safra, N.; Young, A.; Karmi, N.; Schaible, R.S.; Ling, G.V. Mutations in the SLC2A9 Gene Cause Hyperuricosuria and Hyperuricemia in the Dog. PLoS Genet. 2008, 4, e1000246. [Google Scholar] [CrossRef]
- Center, S.A. Chapter 37—The Liver, Biliary Tract, and Exocrine Pancreas. In Small Animal Pediatrics; Peterson, M.E., Kutzler, M.A., Eds.; W.B. Saunders: Saint Louis, MO, USA, 2011; pp. 368–390. ISBN 978-1-4160-4889-3. [Google Scholar]
- Solano-Gallego, L.; Miró, G.; Koutinas, A.; Cardoso, L.; Pennisi, M.G.; Ferrer, L.; Bourdeau, P.; Oliva, G.; Baneth, G. The LeishVet Group, null LeishVet Guidelines for the Practical Management of Canine Leishmaniosis. Parasit. Vectors 2011, 4, 86. [Google Scholar] [CrossRef]
- Gordon, J.M.; Kutzler, M.A. Chapter 38—The Urinary System. In Small Animal Pediatrics; Peterson, M.E., Kutzler, M.A., Eds.; W.B. Saunders: Saint Louis, MO, USA, 2011; pp. 391–404. ISBN 978-1-4160-4889-3. [Google Scholar]
- Torres, M.; Pastor, J.; Roura, X.; Tabar, M.-D.; Espada, Y.; Font, A.; Balasch, J.; Planellas, M. Adverse Urinary Effects of Allopurinol in Dogs with Leishmaniasis: Urinary Adverse Effects of Allopurinol. J. Small Anim. Pract. 2016, 57, 299–304. [Google Scholar] [CrossRef]
- Tas, B.M.; Susenbeth, A. Urinary Purine Derivates Excretion as an Indicator of in Vivo Microbial N Flow in Cattle: A Review. Livest. Sci. 2007, 111, 181–192. [Google Scholar] [CrossRef]
- Moorby, J.M.; Dewhurst, R.J.; Evans, R.T.; Danelón, J.L. Effects of Dairy Cow Diet Forage Proportion on Duodenal Nutrient Supply and Urinary Purine Derivative Excretion. J. Dairy Sci. 2006, 89, 3552–3562. [Google Scholar] [CrossRef]
- Boudra, H.; Doreau, M.; Noziere, P.; Pujos-Guillot, E.; Morgavi, D.P. Simultaneous Analysis of the Main Markers of Nitrogen Status in Dairy Cow’s Urine Using Hydrophilic Interaction Chromatography and Tandem Mass Spectrometry Detection. J. Chromatogr. A 2012, 1256, 169–176. [Google Scholar] [CrossRef]
- Vlassa, M.; Coman, V.; Dragomir, C. Determination of Purine Derivatives in Bovine Urine Using Rapid Chromatographic Techniques. Arch Zootec 2009, 12, 59–70. [Google Scholar]
- Westropp, J.L.; Larsen, J.A.; Johnson, E.G.; Bannasch, D.; Fascetti, A.J.; Biourge, V.; Queau, Y. Evaluation of Dogs with Genetic Hyperuricosuria and Urate Urolithiasis Consuming a Purine Restricted Diet: A Pilot Study. BMC Vet. Res. 2017, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Bartges, J.W.; Osborne, C.A.; Felice, L.J.; Chen, M.; Ulrich, L.K. Effects of Time and Dilution on Concentration of Xanthine in Frozen Urine and Plasma of Dogs. Am. J. Vet. Res. 1997, 58, 118–120. [Google Scholar] [CrossRef]
- Felice, L.J.; Dombrovskis, D.; Lafond, E.; Bartges, J.; Osborne, C.A. Determination of Uric Acid in Canine Serum and Urine by High Performance Liquid Chromatography. Vet. Clin. Pathol. 1990, 19, 86–89. [Google Scholar] [CrossRef]
- George, S.K.; Dipu, M.T.; Mehra, U.R.; Singh, P.; Verma, A.K.; Ramgaokar, J.S. Improved HPLC Method for the Simultaneous Determination of Allantoin, Uric Acid and Creatinine in Cattle Urine. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006, 832, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Li, Y.; Luo, Y.-X.; Li, H.; Gao, X.-F. A New Spectrophotometric Method for Uric Acid Detection Based on Copper Doped Mimic Peroxidase. Anal. Biochem. 2023, 664, 115045. [Google Scholar] [CrossRef]
- Seger, C.; Salzmann, L. After Another Decade: LC-MS/MS Became Routine in Clinical Diagnostics. Clin. Biochem. 2020, 82, 2–11. [Google Scholar] [CrossRef]
- Shipkova, M.; Svinarov, D. LC–MS/MS as a Tool for TDM Services: Where are We? Clin. Biochem. 2016, 49, 1009–1023. [Google Scholar] [CrossRef]
- Viette, V.; Hochstrasser, D.; Fathi, M. LC-MS (/MS) in Clinical Toxicology Screening Methods. Chimia 2012, 66, 339–342. [Google Scholar] [CrossRef]
- Butter, J.J.; Koopmans, R.P.; Michel, M.C. A Rapid and Validated HPLC Method to Quantify Sphingosine 1-Phosphate in Human Plasma Using Solid-Phase Extraction Followed by Derivatization with Fluorescence Detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 824, 65–70. [Google Scholar] [CrossRef]
- Vogeser, M.; Parhofer, K.G. Liquid Chromatography Tandem-Mass Spectrometry (LC-MS/MS)—Technique and Applications in Endocrinology. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2007, 115, 559–570. [Google Scholar] [CrossRef]
- Carvalho, V.M. The Coming of Age of Liquid Chromatography Coupled to Tandem Mass Spectrometry in the Endocrinology Laboratory. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2012, 883–884, 50–58. [Google Scholar] [CrossRef]
- Xie, S.; Lu, Y.; Wang, J.; Lin, C.; Ye, P.; Liu, X.; Xiong, W.; Zeng, Z.; Zeng, D. Development and Validation of an LC–MS/MS Method for the Simultaneous Quantification of Milbemycin Oxime and Praziquantel in Plasma: Application to a Pharmacokinetic Study in Cats. Front. Vet. Sci. 2023, 10, 1285932. [Google Scholar] [CrossRef]
- Bardhi, A.; Vecchiato, C.G.; Sabetti, M.C.; Tardo, A.M.; Vasylyeva, K.; Biagi, G.; Pietra, M.; Barbarossa, A. A Novel UHPLC–MS/MS Method for the Measurement of 25-Hydroxyvitamin D3 in Canine Serum and Its Application to Healthy Dogs. Animals 2024, 14, 62. [Google Scholar] [CrossRef]
- Barbarossa, A.; Bardhi, A.; Gazzotti, T.; Pagliuca, G. A Critical Point in Chiral Chromatography-Mass Spectrometry Analysis of Ketamine Metabolites. Drug Test. Anal. 2021, 13, 1689–1692. [Google Scholar] [CrossRef]
- Bardhi, A.; Romano, J.E.; Pagliuca, G.; Caneschi, A.; Barbarossa, A. Florfenicol and Florfenicol Amine Quantification in Bull Serum and Seminal Plasma by a Single Validated UHPLC-MS/MS Method. Vet. Med. Int. 2023, 2023, 6692920. [Google Scholar] [CrossRef]
- Holst, B.S.; Kushnir, M.M.; Bergquist, J. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for Analysis of Endogenous Steroids in the Luteal Phase and Early Pregnancy in Dogs: A Pilot Study. Vet. Clin. Pathol. 2015, 44, 552–558. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Ghassabian, S.; Moosavi, S.M.; Ghassabian, S. Linearity of Calibration Curves for Analytical Methods: A Review of Criteria for Assessment of Method Reliability. In Calibration and Validation of Analytical Methods—A Sampling of Current Approaches; IntechOpen: London, UK, 2018; ISBN 978-1-78923-085-7. [Google Scholar]
- Nelis, M.; Augustijns, P.; Cabooter, D. Strategies for the Quantification of Endogenously Present Small Molecules in Biological Samples. LC GC Eur. 2019, 32, 354–363. [Google Scholar]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; Alamoudi, J.A.; Alnouti, Y. Quantitative Analysis of Endogenous Compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. [Google Scholar] [CrossRef]
- EMA. ICH M10 on Bioanalytical Method Validation—Scientific Guideline. Available online: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline (accessed on 24 July 2023).
- Pang, S.; Cowen, S. A Generic Standard Additions Based Method to Determine Endogenous Analyte Concentrations by Immunoassays to Overcome Complex Biological Matrix Interference. Sci. Rep. 2017, 7, 17542. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Minakata, K.; Suzuki, M.; Suzuki, O. The Standard Addition Method and Its Validation in Forensic Toxicology. Forensic Toxicol. 2021, 39, 311–333. [Google Scholar] [CrossRef]
- Coglianese, A.; Charlier, B.; Mensitieri, F.; Filippelli, A.; Izzo, V.; Dal Piaz, F. Standard Addition Method (SAM) in LC-MS/MS to Quantify Gluten-Derived Metabolites in Urine Samples. J. Pharm. Biomed. Anal. 2023, 232, 115416. [Google Scholar] [CrossRef] [PubMed]
- Boscher, A.; Guignard, C.; Pellet, T.; Hoffmann, L.; Bohn, T. Development of a Multi-Class Method for the Quantification of Veterinary Drug Residues in Feedingstuffs by Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2010, 1217, 6394–6404. [Google Scholar] [CrossRef]
- van de Merbel, N.C.; Mentink, C.J.A.L.; Hendriks, G.; Wolffenbuttel, B.H.R. Liquid Chromatographic Method for the Quantitative Determination of Nepsilon-Carboxymethyllysine in Human Plasma Proteins. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 808, 163–168. [Google Scholar] [CrossRef]
- Cimetiere, N.; Soutrel, I.; Lemasle, M.; Laplanche, A.; Crocq, A. Standard Addition Method for the Determination of Pharmaceutical Residues in Drinking Water by SPE-LC-MS/MS. Environ. Technol. 2013, 34, 3031–3041. [Google Scholar] [CrossRef]
- Friedrichs, K.R.; Harr, K.E.; Freeman, K.P.; Szladovits, B.; Walton, R.M.; Barnhart, K.F.; Blanco-Chavez, J. ASVCP Reference Interval Guidelines: Determination of de Novo Reference Intervals in Veterinary Species and Other Related Topics. Vet. Clin. Pathol. 2012, 41, 441–453. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Dog Serum | Dog Urine | |||||||
|---|---|---|---|---|---|---|---|---|
| Uric Acid | Allantoin | Uric Acid | Allantoin | |||||
| QC1 (2.5 µg/mL) | QC1 (10 µg/mL) | QC1 (50 µg/mL) | QC1 (500 µg/mL) | |||||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1 (n = 3) | −6.3 | 7.9 | 1.3 | 6.4 | −0.5 | 0.8 | 3.8 | 6.3 |
| 2 (n = 3) | 12.2 | 9.6 | −3.2 | 1.0 | −3.6 | 6.4 | 13.8 | 10.3 |
| 3 (n = 3) | −7.7 | 13.3 | 1.6 | 6.7 | −3.7 | 6.3 | 4.7 | 5.6 |
| Inter-day (n = 9) | −0.6 | 13.2 | −0.1 | 5.1 | −2.6 | 4.7 | 7.4 | 8.2 |
| QC2 (10 µg/mL) | QC2 (50 µg/mL) | QC2 (250 µg/mL) | QC2 (2500 µg/mL) | |||||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1 (n = 3) | −2.1 | 5.1 | 6.7 | 4.7 | 0.1 | 0.4 | 2.6 | 4.4 |
| 2 (n = 3) | −3.9 | 9.2 | 0.3 | 2.8 | −3.6 | 3.2 | 5.4 | 7.3 |
| 3 (n = 3) | −3.6 | 12.4 | −2.2 | 2.9 | −2.6 | 3.4 | 6.6 | 8.1 |
| Inter-day (n = 9) | −3.2 | 7.9 | 1.6 | 5.1 | −2.1 | 2.9 | 4.9 | 6.1 |
| QC3 (50 µg/mL) | QC3 (200 µg/mL) | QC3 (2500 µg/mL) | QC3 (10,000 µg/mL) | |||||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1(n = 3) | 4.7 | 2.1 | 4.4 | 4.6 | 2.9 | 7.6 | 5.4 | 5.8 |
| 2 (n = 3) | 2.3 | 3.4 | −0.8 | 1.1 | −10.9 | 14.1 | 5.0 | 6.1 |
| 3 (n = 3) | 0.7 | 4.8 | −2.1 | 2.2 | −8.0 | 14.6 | 5.5 | 9.7 |
| Inter-day (n = 9) | 2.6 | 3.5 | 0.5 | 4.3 | −5.3 | 12.5 | 5.3 | 6.4 |
| Bovine Urine | ||||
|---|---|---|---|---|
| Uric Acid | Allantoin | |||
| QC1 (50 µg/mL) | QC1 (500 µg/mL) | |||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1 (n = 3) | −5.9 | 5.5 | −8.3 | 7.1 |
| 2 (n = 3) | 0.6 | 6.6 | −8.4 | 5.8 |
| 3 (n = 3) | 0.0 | 13.2 | −5.6 | 1.5 |
| Inter-day (n = 9) | −1.8 | 8.6 | −7.4 | 4.8 |
| QC2 (250 µg/mL) | QC2 (2500 µg/mL) | |||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1 (n = 3) | −3.6 | 3.3 | −0.8 | 2.0 |
| 2 (n = 3) | −3.7 | 10.1 | −4.9 | 3.2 |
| 3 (n = 3) | 1.5 | 7.1 | −1.4 | 1.9 |
| Inter-day (n = 9) | −1.9 | 6.9 | −2.4 | 2.8 |
| QC3 (2500 µg/mL) | QC3 (10,000 µg/mL) | |||
| Day | Accuracy (%) | Precision (CV%) | Accuracy (%) | Precision (CV%) |
| 1 (n = 3) | −5.1 | 7.2 | 0.1 | 1.9 |
| 2 (n = 3) | −0.4 | 3.7 | 0.5 | 4.5 |
| 3 (n = 3) | −1.6 | 1.4 | 0.4 | 4.6 |
| Inter-day (n = 9) | −2.4 | 4.6 | 0.3 | 3.4 |
| Serum | Standard Addition | Water | Background Subtraction | ||
|---|---|---|---|---|---|
| Uric Acid (µg/mL) | |||||
| Dog 1 | 3.3 | 1.6 (−51%) | 2.3 (−29%) | 2.3 (−29%) | 2.5 (−23%) |
| Dog 2 | 3.9 | 2.7 (−31%) | 3.7 (−6%) | 4.0 (+2%) | 6.9 (+76%) |
| Dog 3 | 4.9 | 3.4 (−30%) | 4.9 (+1%) | 5.1 (+5%) | 8.7 (+79%) |
| Dog 4 | 4.6 | 2.9 (−36%) | 4.1 (−10%) | 4.1 (−10%) | 7.5 (+65%) |
| Allantoin (µg/mL) | |||||
| Dog 1 | 14.9 | 12.5 (−16%) | 19.4 (+30%) | 19.9 (+33%) | 22.6 (+51%) |
| Dog 2 | 22.8 | 12.2 (−47%) | 19.3 (−15%) | 21.9 (−4%) | 17.2 (−25%) |
| Dog 3 | 20.8 | 13.1 (−37%) | 19.9 (−5%) | 23.2 (+11%) | 18.4 (−12%) |
| Dog 4 | 18.9 | 10.4 (−45%) | 16.5 (−13%) | 16.3 (−14%) | 14.7 (−22%) |
| Urine | Standard Addition | Water | Background Subtraction | ||
| Uric Acid (µg/mL) | |||||
| Dog 1 | 530.2 | 672.9 (+27%) | 650.2 (+23%) | 737.1 (+39%) | 653.1 (+23%) |
| Dog 2 | 535.4 | 473.6 (−11%) | 562.6 (+5%) | 460.3 (−14%) | 460.0 (−14%) |
| Dog 3 | 199.2 | 22.0 (−89%) | 287.0 (+44%) | 244.2 (+23%) | 264.2 (+33%) |
| Dog 4 | 76.0 | 100.3 (+32%) | 118.4 (+56%) | 97.0 (+28%) | 109.2 (+44%) |
| Allantoin (µg/mL) | |||||
| Dog 1 | 7276.8 | 7916.0 (+9%) | 8543.1 (+17%) | 7528.2 (+3%) | 5979.0 (−18%) |
| Dog 2 | 8967.1 | 7090.4 (−21%) | 6992.3 (−22%) | 5358.2 (−40%) | 8543.0 (−5%) |
| Dog 3 | 5087.3 | 5101.3 (0%) | 5027.4 (−1%) | 3854.4 (−24%) | 5027.4 (−1%) |
| Dog 4 | 1871.7 | 2673.0 (+43%) | 2633.7 (+41%) | 2873.0 (+53%) | 2542.0 (+36%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bardhi, A.; Dondi, F.; Barbarossa, A. Exploring the Most Effective Strategy for Purine Metabolite Quantification in Veterinary Medicine Using LC–MS/MS. Vet. Sci. 2025, 12, 230. https://doi.org/10.3390/vetsci12030230
Bardhi A, Dondi F, Barbarossa A. Exploring the Most Effective Strategy for Purine Metabolite Quantification in Veterinary Medicine Using LC–MS/MS. Veterinary Sciences. 2025; 12(3):230. https://doi.org/10.3390/vetsci12030230
Chicago/Turabian StyleBardhi, Anisa, Francesco Dondi, and Andrea Barbarossa. 2025. "Exploring the Most Effective Strategy for Purine Metabolite Quantification in Veterinary Medicine Using LC–MS/MS" Veterinary Sciences 12, no. 3: 230. https://doi.org/10.3390/vetsci12030230
APA StyleBardhi, A., Dondi, F., & Barbarossa, A. (2025). Exploring the Most Effective Strategy for Purine Metabolite Quantification in Veterinary Medicine Using LC–MS/MS. Veterinary Sciences, 12(3), 230. https://doi.org/10.3390/vetsci12030230