

Whole-Genome Analysis of Porcine Epidemic Diarrhea Virus from Yunnan, China

Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection and PEDV Detection
2.2. Sequence Assembly and Alignment of Whole Genome
2.3. Database Accession Numbers
2.4. Phylogenetic Analysis of S Gene and PEDV Genome
2.5. Genetic Recombination Analysis
3. Results
3.1. Epidemiology of PEDV in Yunnan from February 2021 to December 2023
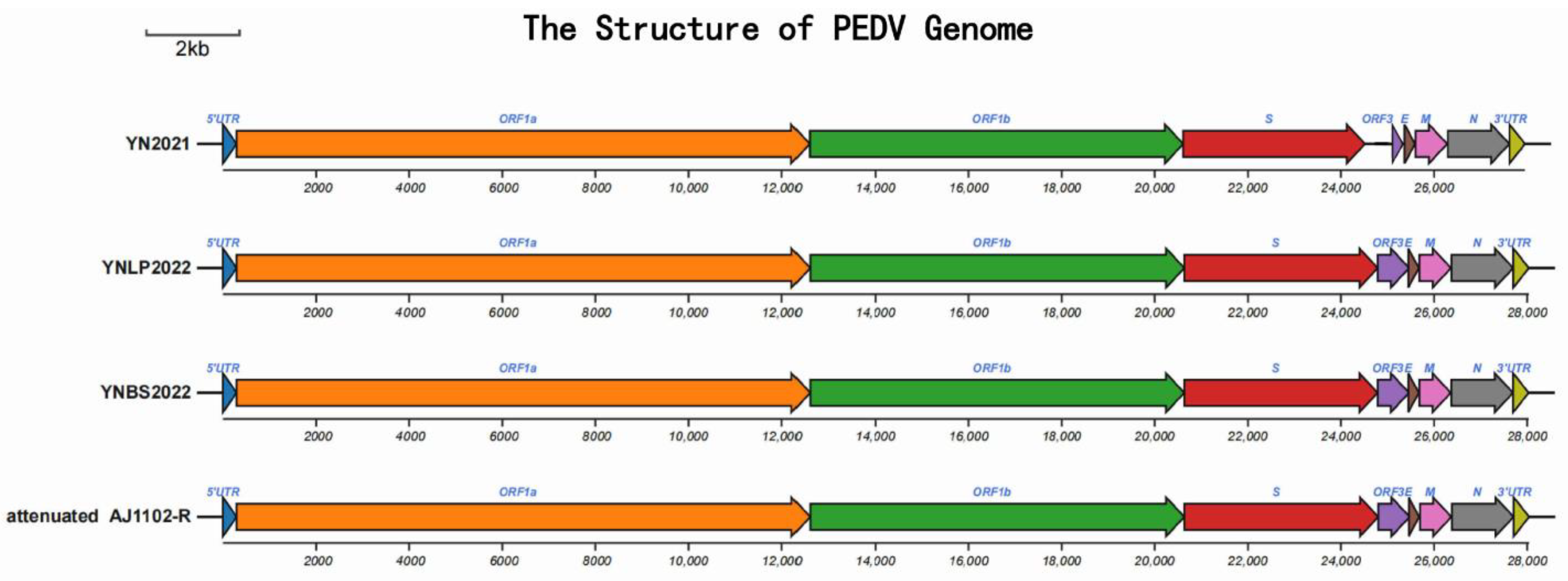
3.2. Genome Information of Yunnan PEDV Strains
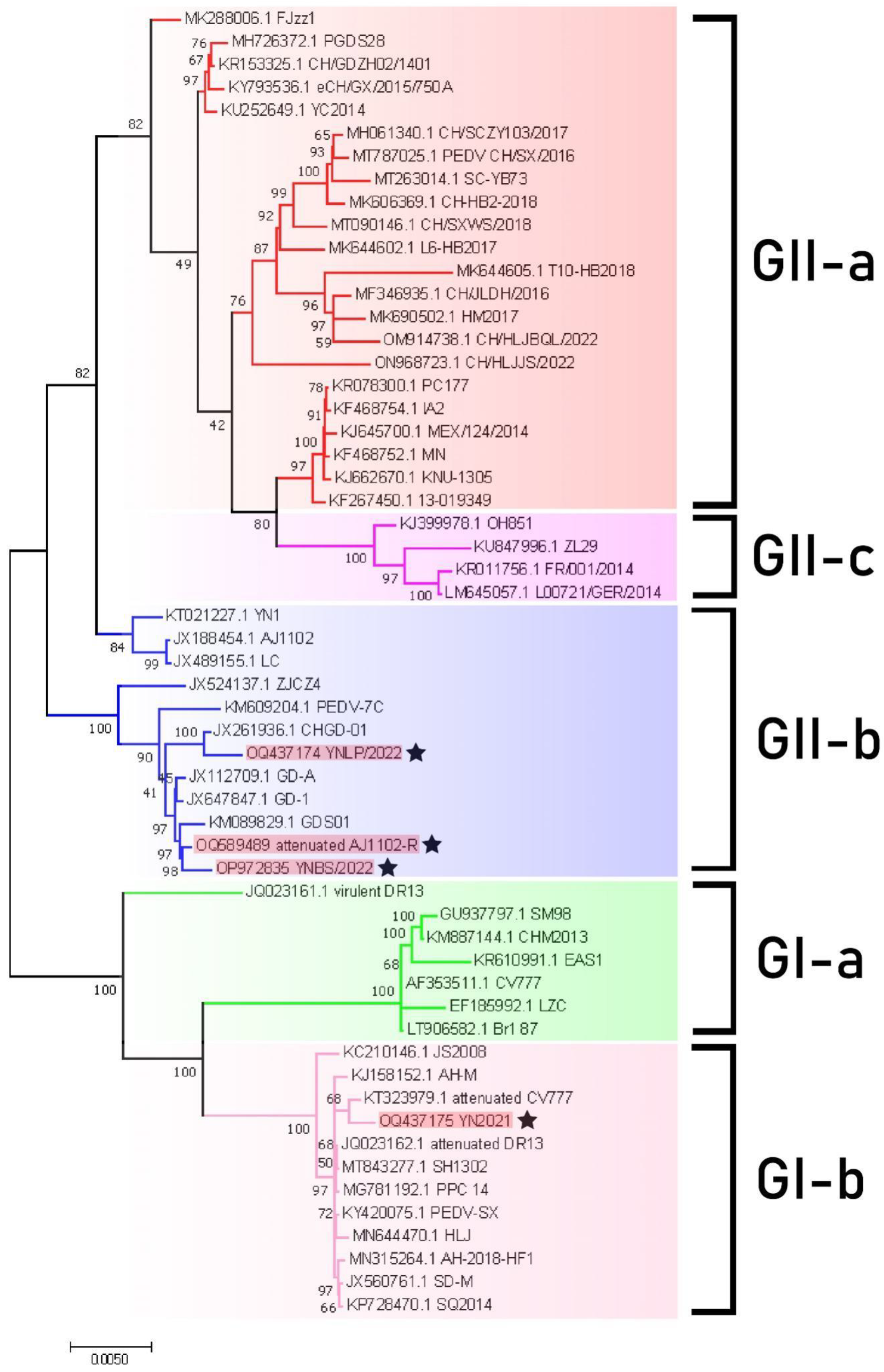
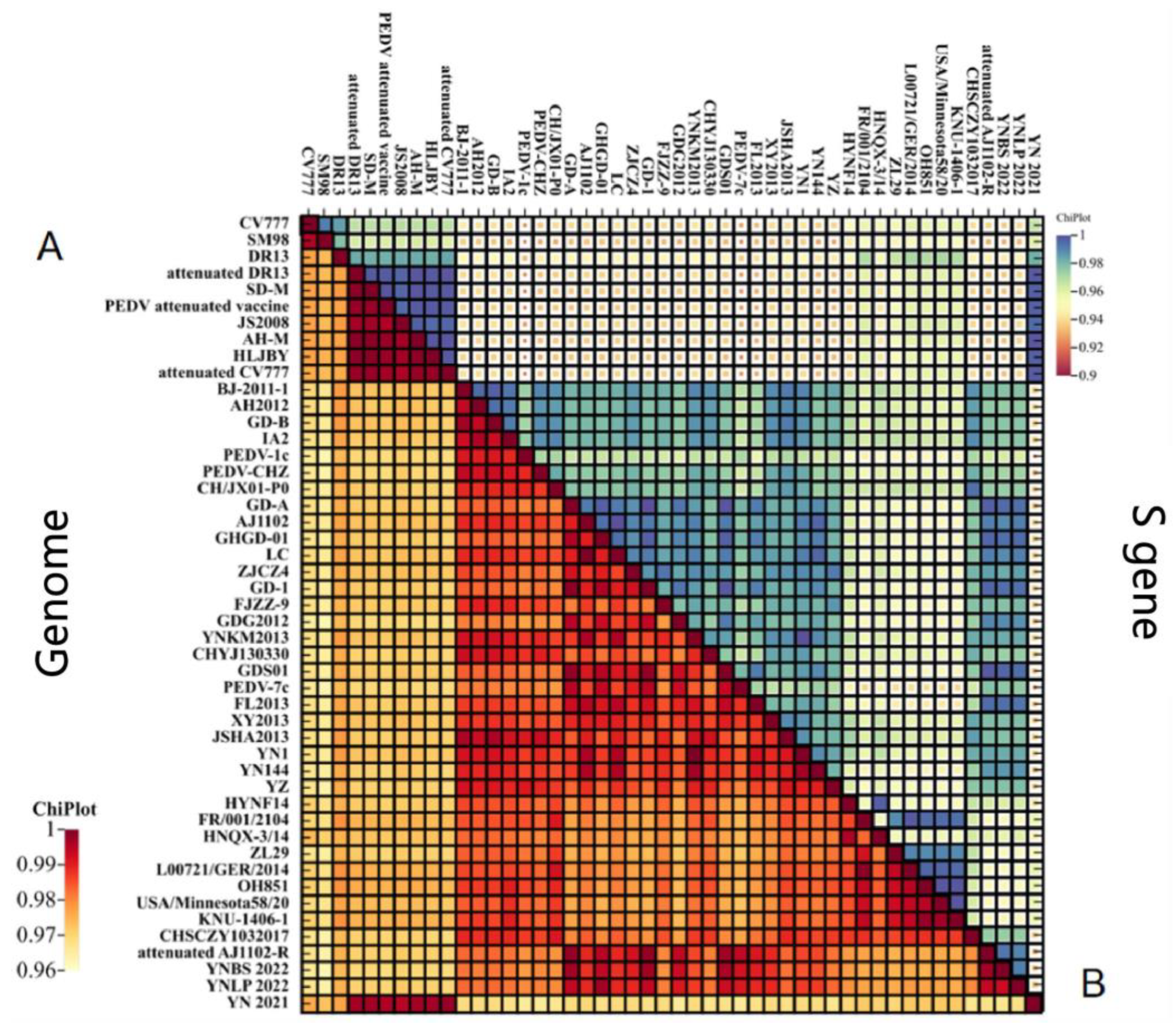
3.3. Phylogenetic Analysis of PEDV Genome
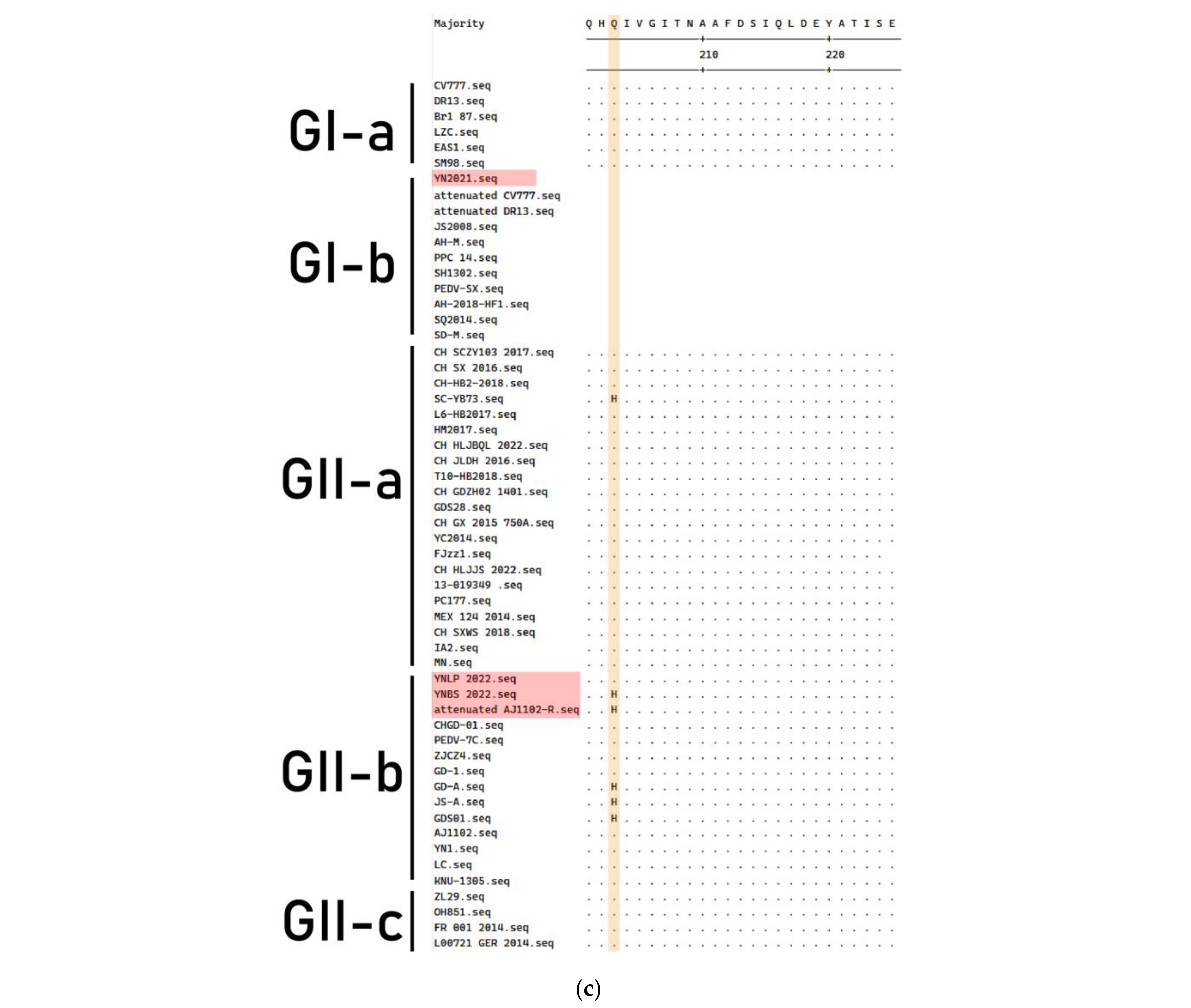
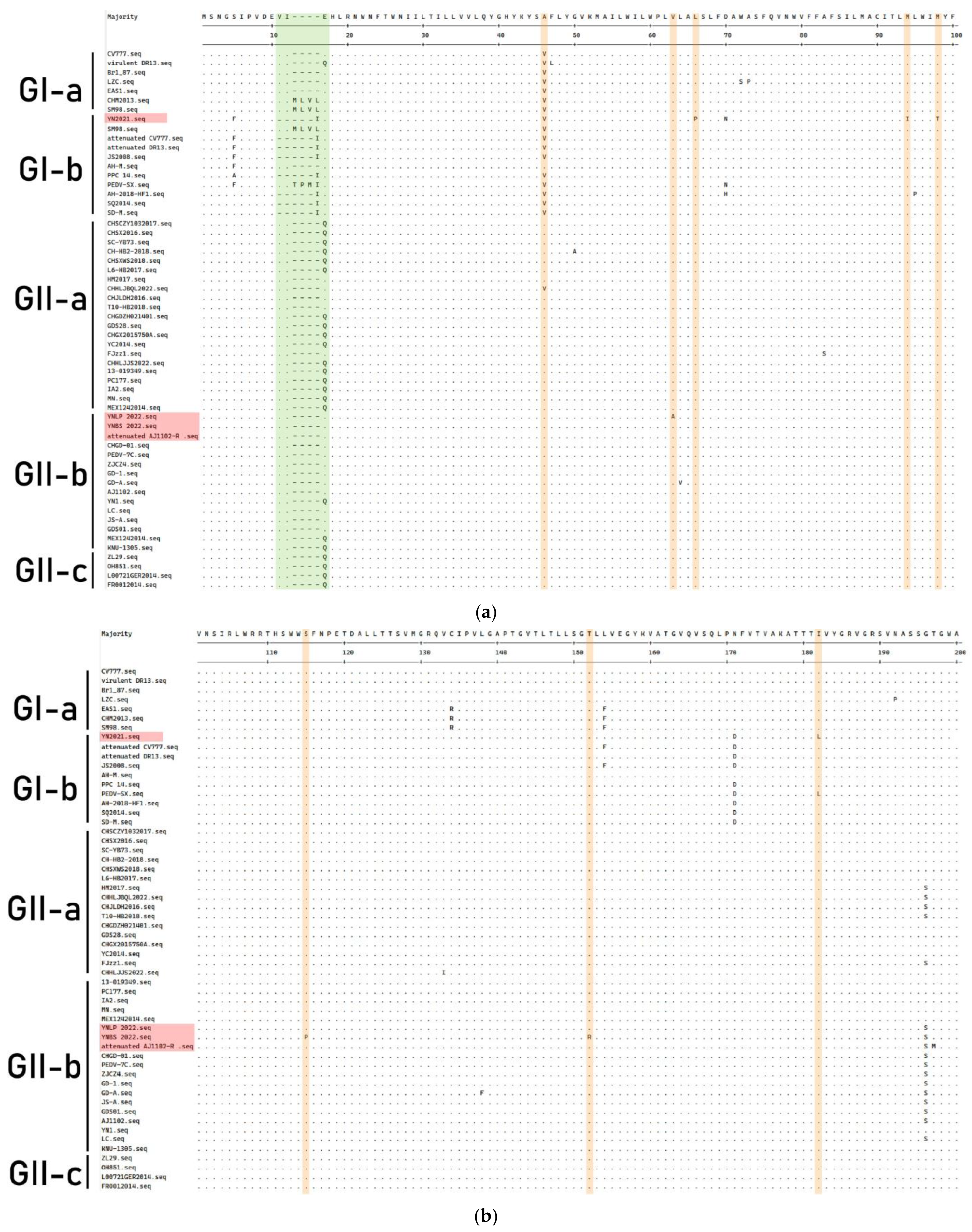
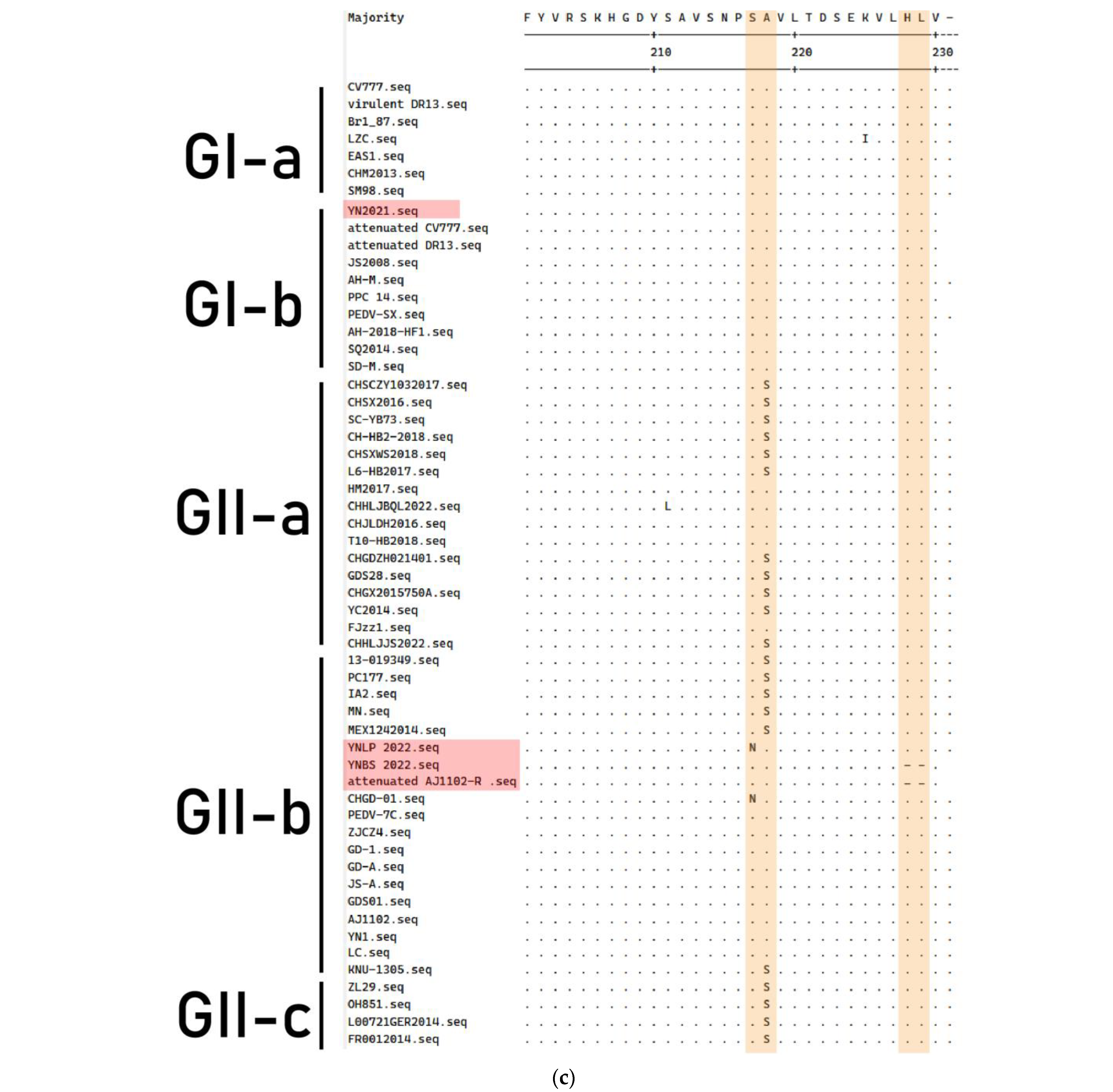
3.4. Genetic Variation of S Gene
3.5. Genetic Variation of ORF3 Gene
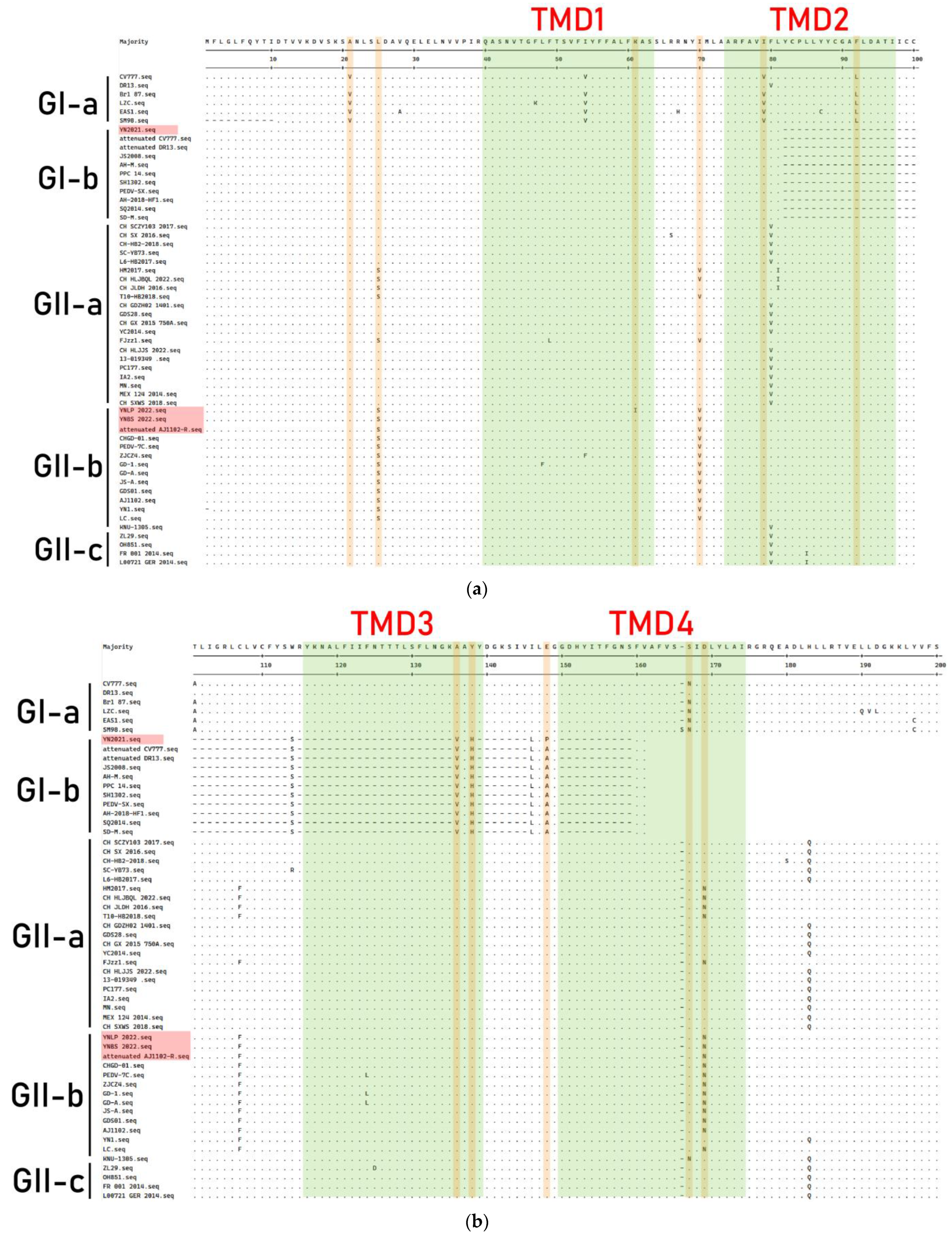
3.6. Genetic Variation of M Gene
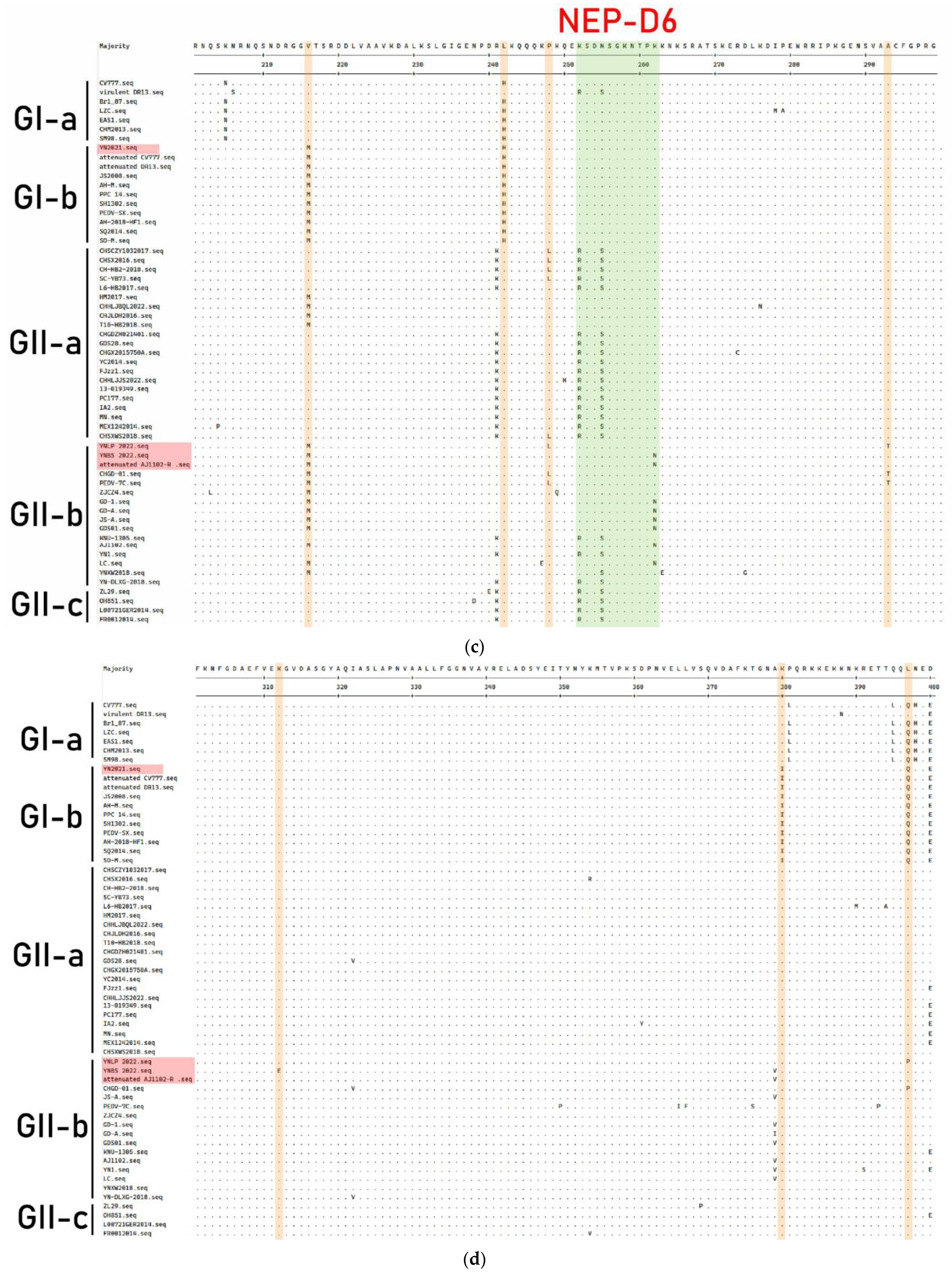
3.7. Genetic Variation of N Protein
3.8. Genome-Wide Recombination Analysis of PEDV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Res. 2020, 286, 198045. [Google Scholar] [CrossRef] [PubMed]
- Chasey, D.; Cartwright, S.F. Virus-like particles associated with porcine epidemic diarrhoea. Res. Vet. Sci. 1978, 25, 255–256. [Google Scholar] [CrossRef]
- Lin, C.M.; Saif, L.J.; Marthaler, D.; Wang, Q. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res. 2016, 226, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Xiao, S. Porcine epidemic diarrhea in China. Virus Res. 2016, 226, 7–13. [Google Scholar] [CrossRef]
- Sun, M.; Ma, J.; Wang, Y.; Wang, M.; Song, W.; Zhang, W.; Lu, C.; Yao, H. Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. J. Clin. Microbiol. 2015, 53, 1484–1492. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Shi, H.; Qiu, H.J.; Liu, S.; Shi, D.; Zhang, X.; Feng, L. Complete genome sequence of a Chinese virulent porcine epidemic diarrhea virus strain. J. Virol. 2011, 85, 11538–11539. [Google Scholar] [CrossRef]
- Tan, L.; Li, Y.; He, J.; Hu, Y.; Cai, X.; Liu, W.; Liu, T.; Wang, J.; Li, Z.; Yuan, X.; et al. Epidemic and genetic characterization of porcine epidemic diarrhea virus strains circulating in the regions around Hunan, China, during 2017–2018. Arch. Virol. 2020, 165, 877–889. [Google Scholar] [CrossRef]
- Su, M.; Li, C.; Qi, S.; Yang, D.; Jiang, N.; Yin, B.; Guo, D.; Kong, F.; Yuan, D.; Feng, L.; et al. A molecular epidemiological investigation of PEDV in China: Characterization of co-infection and genetic diversity of S1-based genes. Transbound. Emerg. Dis. 2020, 67, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Niu, B.B.; Yan, H.; Gao, D.S.; Yang, X.; Chen, L.; Chang, H.T.; Zhao, J.; Wang, C.Q. Genetic properties of endemic Chinese porcine epidemic diarrhea virus strains isolated since 2010. Arch Virol. 2013, 158, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.Q.; Cai, R.J.; Chen, Y.Q.; Liang, P.S.; Chen, D.K.; Song, C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012, 18, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, Y.; Han, X.; Yu, Z.; Wei, Y.; Zhang, G. Porcine epidemic diarrhea virus in Asia: An alarming threat to the global pig industry. Infect. Genet. Evol. 2019, 70, 24–26. [Google Scholar] [CrossRef]
- Kocherhans, R.; Bridgen, A.; Ackermann, M.; Tobler, K. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001, 23, 137–144. [Google Scholar] [CrossRef]
- Duarte, M.; Gelfi, J.; Lambert, P.; Rasschaert, D.; Laude, H. Genome organization of porcine epidemic diarrhoea virus. Adv. Exp. Med. Biol. 1993, 342, 55–60. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [CrossRef]
- Guo, J.; Fang, L.; Ye, X.; Chen, J.; Xu, S.; Zhu, X.; Miao, Y.; Wang, D.; Xiao, S. Evolutionary and genotypic analyses of global porcine epidemic diarrhea virus strains. Transbound. Emerg. Dis. 2019, 66, 111–118. [Google Scholar] [CrossRef]
- Pensaert, M.B.; Martelli, P. Porcine epidemic diarrhea: A retrospect from Europe and matters of debate. Virus Res. 2016, 226, 1–6. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, M.; Zhan, J.; Liu, Q.Y.; Fang, L.L.; Zhao, C.Y.; Jiang, P.; Li, Y.F.; Bai, J. Pathogenicity and immunogenicity of a new strain of porcine epidemic diarrhea virus containing a novel deletion in the N gene. Vet. Microbiol. 2020, 240, 108511. [Google Scholar] [CrossRef]
- Fan, B.; Jiao, D.; Zhang, R.; Zhou, J.; Guo, R.; Yu, Z.; Shi, D.; Zhao, Y.; Gu, J.; Niu, B.; et al. Origin and epidemic status of porcine epidemic diarrhea virus variants in China. Transbound. Emerg. Dis. 2020, 67, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, J.R.; Hakze-van der Honing, R.; Almeida, A.; Lourenco, M.; van der Poel, W.H.; Nascimento, M.S. Outbreak of Porcine Epidemic Diarrhea Virus in Portugal, 2015. Transbound. Emerg. Dis. 2015, 62, 586–588. [Google Scholar] [CrossRef]
- Grasland, B.; Bigault, L.; Bernard, C.; Quenault, H.; Toulouse, O.; Fablet, C.; Rose, N.; Touzain, F.; Blanchard, Y. Complete genome sequence of a porcine epidemic diarrhea S gene indel strain isolated in France in December 2014. Genome. Announc. 2015, 3, 10-1128. [Google Scholar] [CrossRef]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg. Infect. Dis. 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Oka, T.; Saif, L.J.; Marthaler, D.; Esseili, M.A.; Meulia, T.; Lin, C.M.; Vlasova, A.N.; Jung, K.; Zhang, Y.; Wang, Q. Cell culture isolation and sequence analysis of genetically diverse US porcine epidemic diarrhea virus strains including a novel strain with a large deletion in the spike gene. Vet. Microbiol. 2014, 173, 258–269. [Google Scholar] [CrossRef]
- Lei, J.L.; Miao, Y.Q.; Guan, Z.; Chen, H.; Xiang, C.H.; Lu, H.Q.; Fang, Y.; Han, Y.; Hu, R.C.; Lu, K.J.; et al. A porcine epidemic diarrhea virus isolated from a sow farm vaccinated with CV777 strain in Yinchuan, China: Characterization, antigenicity, and pathogenicity. Transbound. Emerg. Dis. 2023, 2023, 7082352. [Google Scholar] [CrossRef]
- Li, W.; Li, H.; Liu, Y.; Pan, Y.; Deng, F.; Song, Y.; Tang, X.; He, Q. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg. Infect. Dis. 2012, 18, 1350–1353. [Google Scholar] [CrossRef]
- He, W.T.; Bollen, N.; Xu, Y.; Zhao, J.; Dellicour, S.; Yan, Z.; Gong, W.; Zhang, C.; Zhang, L.; Lu, M.; et al. Phylogeography Reveals Association between Swine Trade and the spread of porcine epidemic diarrhea virus in China and across the world. Mol. Biol. Evol. 2022, 39, msab364. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Zhou, J.; Wang, X.; Ma, L.; Li, J.; Yang, L.; Yuan, H.; Pang, D.; Ouyang, H. Porcine epidemic diarrhea virus: An ppdated overview of virus epidemiology, virulence variation patterns and virus-host interactions. Viruses 2022, 14, 2434. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Zhao, R.; Liu, J.; Zhao, Q.; Zhu, L.; Zhang, B.; Bi, J.; Yang, G.; Liu, J.; Yin, G. Epidemiology and phylogeny of spike gene of porcine epidemic diarrhea virus from Yunnan, China. Virus Res. 2018, 249, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chai, X.; Cheng, Y.; Xing, G.; Liao, A.; Du, L.; Wang, Y.; Lei, J.; Gu, J.; Zhou, J. Molecular characteristics of the spike gene of porcine epidemic diarrhoea virus strains in Eastern China in 2016. Virus Res. 2018, 247, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Shi, D.; Shi, H.; Zhang, X.; Feng, L. Complete genome sequence of a porcine epidemic diarrhea virus variant. J. Virol. 2012, 86, 3408. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, X.; Li, H.; Ma, B.; Guan, R.; Yang, J.; Chen, D.; Han, X.; Zhou, L.; Song, Z.; et al. Molecular characterization of porcine epidemic diarrhea virus associated with outbreaks in southwest China during 2014–2018. Transbound. Emerg. Dis. 2021, 68, 3482–3497. [Google Scholar] [CrossRef]
- Van Diep, N.; Choijookhuu, N.; Fuke, N.; Myint, O.; Izzati, U.Z.; Suwanruengsri, M.; Hishikawa, Y.; Yamaguchi, R. New tropisms of porcine epidemic diarrhoea virus (PEDV) in pigs naturally coinfected by variants bearing large deletions in the spike (S) protein and PEDVs possessing an intact S protein. Transbound. Emerg. Dis. 2020, 67, 2589–2601. [Google Scholar] [CrossRef]
- Crawford, K.; Lager, K.; Miller, L.; Opriessnig, T.; Gerber, P.; Hesse, R. Evaluation of porcine epidemic diarrhea virus transmission and the immune response in growing pigs. Vet. Res. 2015, 46, 49. [Google Scholar] [CrossRef]
- Song, C.; Liu, N.; Li, M.; Shi, Y.; Li, H.; Gao, L.; Yang, R.; Yao, J. Sequence Analysis of N Gene of Porcine Epidemic Diarrhea Virus Epidemic Strains in Yunnan Province. Prog. Vet. Med. 2016, 37, 5. [Google Scholar] [CrossRef]
- Liang, W.; Zhou, D.; Geng, C.; Yang, K.; Duan, Z.; Guo, R.; Liu, W.; Yuan, F.; Liu, Z.; Gao, T.; et al. Isolation and evolutionary analyses of porcine epidemic diarrhea virus in Asia. PeerJ 2020, 8, e10114. [Google Scholar] [CrossRef]
- Zhang, H.; Han, F.; Yan, X.; Liu, L.; Shu, X.; Hu, H. Prevalence and phylogenetic analysis of spike gene of porcine epidemic diarrhea virus in Henan province, China in 2015–2019. Infect. Genet. Evol. 2021, 88, 104709. [Google Scholar] [CrossRef]
- Lara-Romero, R.; Gomez-Nunez, L.; Cerriteno-Sanchez, J.L.; Marquez-Valdelamar, L.; Mendoza-Elvira, S.; Ramirez-Mendoza, H.; Rivera-Benitez, J.F. Molecular characterization of the spike gene of the porcine epidemic diarrhea virus in Mexico, 2013–2016. Virus Genes 2018, 54, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Son, K.Y.; Noh, Y.H.; Lee, S.C.; Choi, H.W.; Yoon, I.J.; Lee, C. Genetic characteristics, pathogenicity, and immunogenicity associated with cell adaptation of a virulent genotype 2b porcine epidemic diarrhea virus. Vet. Microbiol. 2017, 207, 248–258. [Google Scholar] [CrossRef]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H., 3rd; Leist, S.R.; Yount, B.L., Jr.; McAnarney, E.T.; Graham, R.L.; Waters, K.M.; Baric, R.S. Combination Attenuation Offers Strategy for Live Attenuated Coronavirus Vaccines. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Si, F.; Chen, B.; Hu, X.; Yu, R.; Dong, S.; Wang, R.; Li, Z. Porcine Epidemic Diarrhea Virus ORF3 Protein Is Transported through the Exocytic Pathway. J. Virol. 2020, 94, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lu, W.; Chen, J.; Xie, S.; Shi, H.; Hsu, H.; Yu, W.; Xu, K.; Bian, C.; Fischer, W.B.; et al. PEDV ORF3 encodes an ion channel protein and regulates virus production. FEBS Lett. 2012, 586, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Zhu, L.; Ma, J.Y.; Zhou, Q.F.; Song, Y.H.; Sun, B.L.; Chen, R.A.; Xie, Q.M.; Bee, Y.Z. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field strains in south China. Virus Genes. 2012, 45, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Olech, M.; Antas, M.; Szczotka-Bochniarz, A. Characterisation of ORF3, M, N and E Gene Sequences of Porcine Epidemic Diarrhoea Virus from Domestic Pigs in Poland. J. Vet. Res. 2022, 66, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Leng, Z.; Zhai, S.L.; Chen, D.; Song, C. Genetic variability and phylogeny of current Chinese porcine epidemic diarrhea virus strains based on spike, ORF3, and membrane genes. Sci. World J. 2014, 2014, 208439. [Google Scholar] [CrossRef]
- Kim, S.J.; Nguyen, V.G.; Huynh, T.M.; Park, Y.H.; Park, B.K.; Chung, H.C. Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene. Viruses 2020, 12, 790. [Google Scholar] [CrossRef]
- Wang, K.; Xie, C.; Zhang, J.; Zhang, W.; Yang, D.; Yu, L.; Jiang, Y.; Yang, S.; Gao, F.; Yang, Z.; et al. The Identification and Characterization of Two Novel Epitopes on the Nucleocapsid Protein of the Porcine Epidemic Diarrhea Virus. Sci. Rep. 2016, 6, 39010. [Google Scholar] [CrossRef]
- Vieth, S.; Torda, A.E.; Asper, M.; Schmitz, H.; Gunther, S. Sequence analysis of L RNA of Lassa virus. Virology 2004, 318, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Rabadan, R.; Levine, A.J.; Krasnitz, M. Non-random reassortment in human influenza A viruses. Influenza Other Respir. Viruses 2008, 2, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Iturriza-Gomara, M.; Isherwood, B.; Desselberger, U.; Gray, J. Reassortment in vivo: Driving force for diversity of human rotavirus strains isolated in the United Kingdom between 1995 and 1999. J. Virol. 2001, 75, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.F.; Jin, Y.L.; Xing, G.; Qv, L.L.; Huang, Y.W.; Zhou, J.Y. Evidence of recombinant strains of porcine epidemic diarrhea virus, United States, 2013. Emerg. Infect. Dis. 2014, 20, 1735–1738. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Sample Numbers | Sample Types | Positive Numbers | Positive Rate (%) | ||
|---|---|---|---|---|---|---|
| Feces | Intestine | Blood | ||||
| 2021 | 128 | 58 | 26 | 44 | 33 | 25.78 |
| 2022 | 84 | 32 | 22 | 30 | 12 | 14.28 |
| 2023 | 7 | 3 | 2 | 2 | 2 | 28.57 |
| Total | 219 | 93 | 50 | 76 | 47 | 21.46 |
| Strain | Length (bp) | ORF1ab (bp) | S (bp) | ORF3 (bp) | E (bp) | M (bp) | N (bp) |
|---|---|---|---|---|---|---|---|
| YN2021 | 27,953 | 20,320 | 4149 | 276 | 231 | 681 | 1326 |
| YNLP2022 | 28,036 | 20,345 | 4158 | 675 | 231 | 681 | 1326 |
| YNBS2022 | 28,031 | 20,345 | 4158 | 675 | 231 | 678 | 1326 |
| AJ1102-R | 28,042 | 20,345 | 4170 | 675 | 231 | 678 | 1326 |
| Strain | Amino Acid Variations Based on CV777 | ||||||
|---|---|---|---|---|---|---|---|
| 577 | 897 | 901 | 982 | 1249 | 1300 | 1301 | |
| CV777 | Q | G | Q | P | D | E | Q |
| DR13 | Q | G | Q | P | D | E | Q |
| Attenuated CV777 | Q | G | Q | P | D | E | Q |
| Attenuated DR13 | Q | G | Q | P | D | E | Q |
| AJ1102 | Q | G | Q | P | D | E | Q |
| Attenuated AJ1102-R | Q | G | Q | P | E | E | Q |
| ZL29 | Q | G | Q | P | D | E | Q |
| YN2021 | R | G | Q | P | D | E | Q |
| YNLP 2022 | Q | G | Q | P | D | E | Q |
| YNBS 2022 | Q | R | H | A | E | Q | H |
| Strain | Detect Method | Recombination Signal Yes/No | p-Value |
|---|---|---|---|
| YN2021 | RDP | − | None |
| GENECONV | − | None | |
| Bootscan | − | None | |
| MaxChi | + | 1.53 × 10−2 | |
| Chimaera | − | None | |
| 3Seq | + | 2.161 × 10−2 | |
| SiSscan | − | None | |
| YNLP 2022 | RDP | + | 1.549 × 10−18 |
| GENECONV | + | 1.961 × 10−18 | |
| Bootscan | + | 4.224 × 10−12 | |
| MaxChi | + | 1.445 × 10−8 | |
| Chimaera | + | 4.310 × 10−9 | |
| 3Seq | + | 3.080 × 10−26 | |
| SiSscan | + | 3.106 × 10−8 | |
| YNBS 2022 | RDP | + | 7.149 × 10−17 |
| GENECONV | + | 6.743 × 10−10 | |
| Bootscan | − | None | |
| MaxChi | + | 1.986 × 10−9 | |
| Chimaera | + | 1.270 × 10−9 | |
| 3Seq | + | 1.439 × 10−7 | |
| SiSscan | + | 3.683 × 10−15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Yin, G.; Wang, Y.; Li, Y.; Wang, X.; Bi, J.; Yang, G.; Qu, K.; Gao, L. Whole-Genome Analysis of Porcine Epidemic Diarrhea Virus from Yunnan, China. Vet. Sci. 2024, 11, 548. https://doi.org/10.3390/vetsci11110548
Zhang R, Yin G, Wang Y, Li Y, Wang X, Bi J, Yang G, Qu K, Gao L. Whole-Genome Analysis of Porcine Epidemic Diarrhea Virus from Yunnan, China. Veterinary Sciences. 2024; 11(11):548. https://doi.org/10.3390/vetsci11110548
Chicago/Turabian StyleZhang, Runting, Gefen Yin, Yunhua Wang, Yongneng Li, Xinxian Wang, Junlong Bi, Guishu Yang, Kaixing Qu, and Libo Gao. 2024. "Whole-Genome Analysis of Porcine Epidemic Diarrhea Virus from Yunnan, China" Veterinary Sciences 11, no. 11: 548. https://doi.org/10.3390/vetsci11110548
APA StyleZhang, R., Yin, G., Wang, Y., Li, Y., Wang, X., Bi, J., Yang, G., Qu, K., & Gao, L. (2024). Whole-Genome Analysis of Porcine Epidemic Diarrhea Virus from Yunnan, China. Veterinary Sciences, 11(11), 548. https://doi.org/10.3390/vetsci11110548