
Microbiome Responses to Oral Fecal Microbiota Transplantation in a Cohort of Domestic Dogs

 ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. FMT Participants
2.2. Preparation of FMT Capsules
2.3. DNA Extractions and Illumina 16S rRNA Gene Sequencing
2.4. Amplicon Sequence Processing in DADA2
2.5. Statistical Analysis of Microbiome Data
3. Results
3.1. Description of Dog Cohort
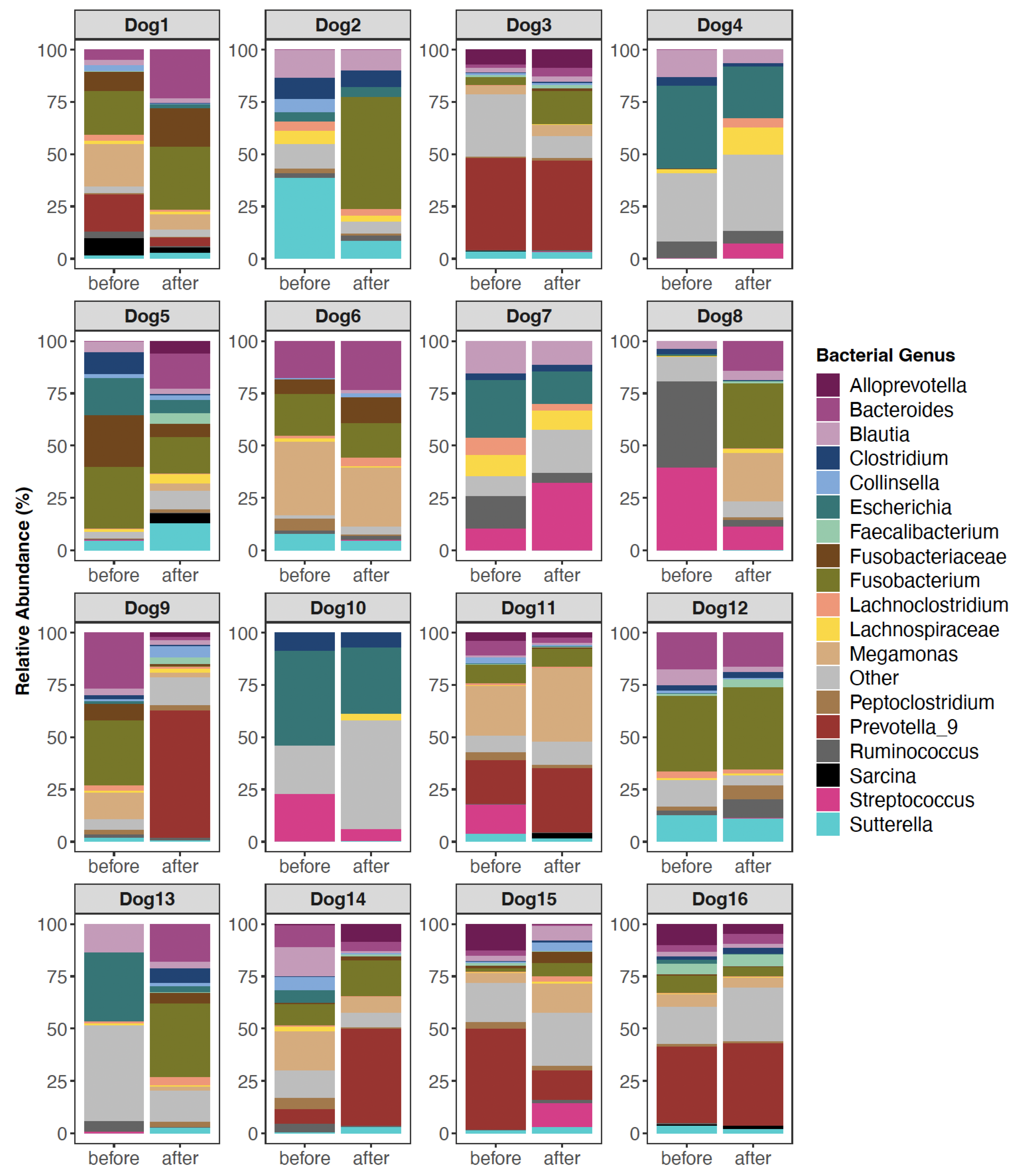
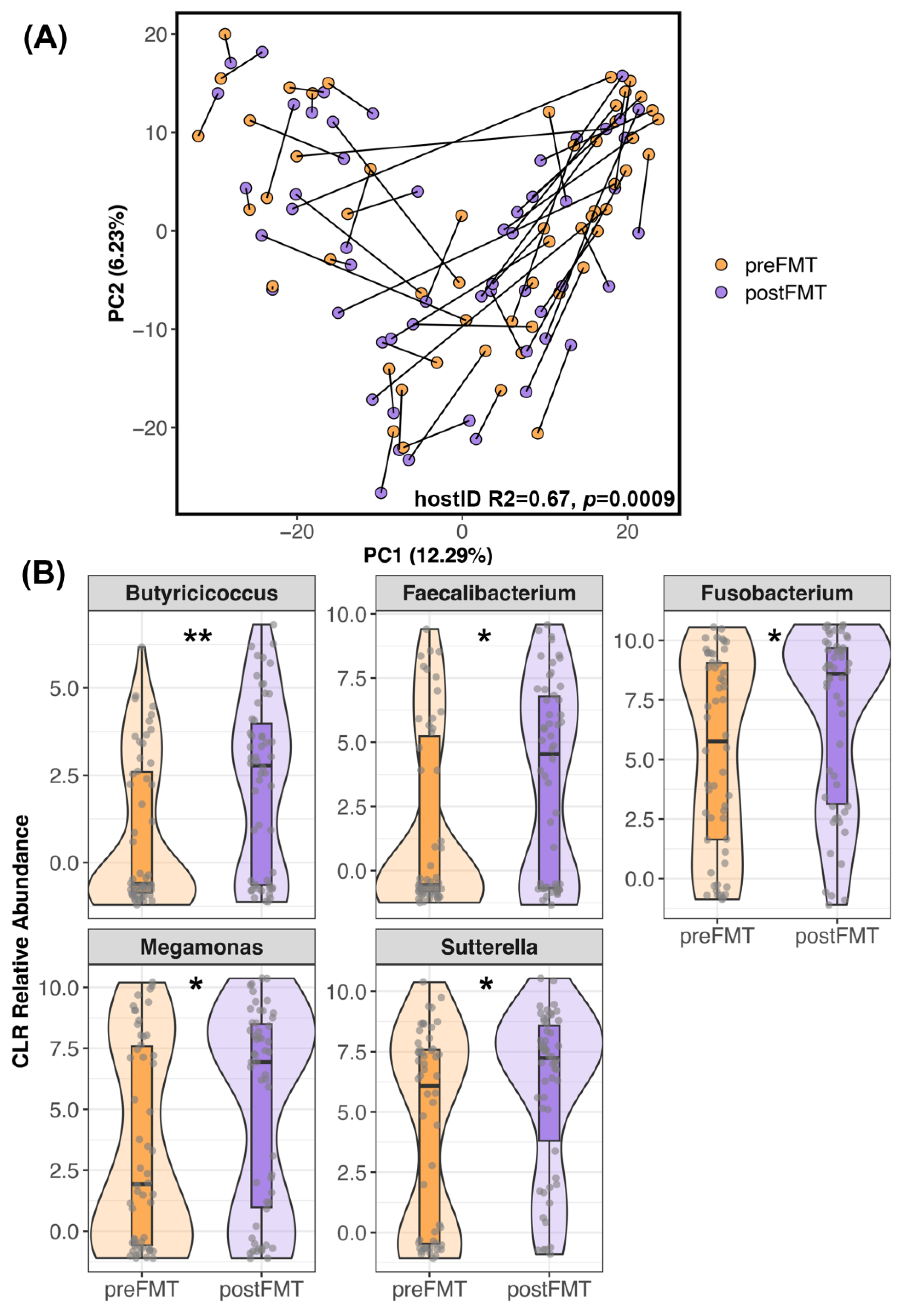
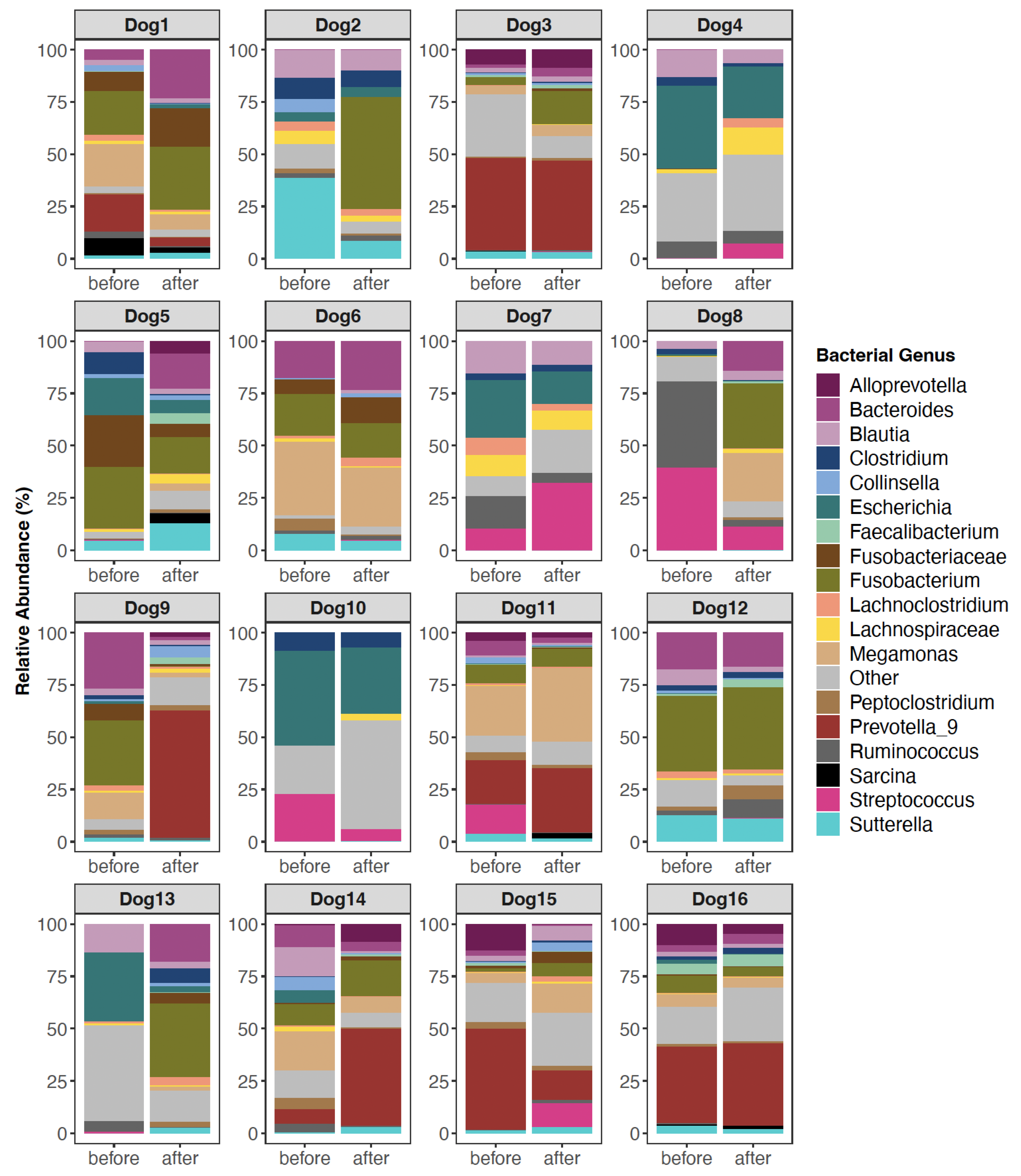
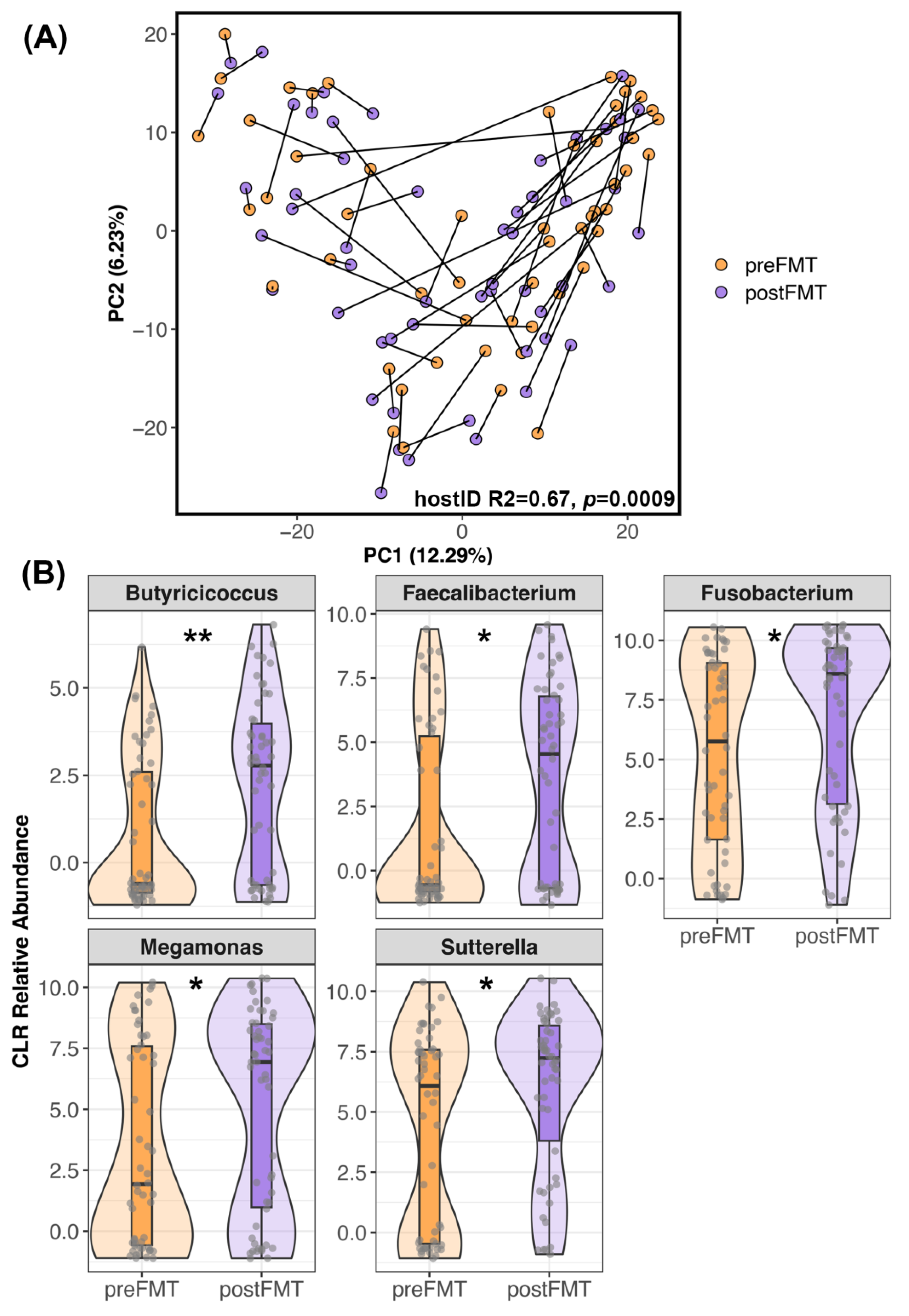
3.2. The Composition of Canine Fecal Microbiomes before and after FMT
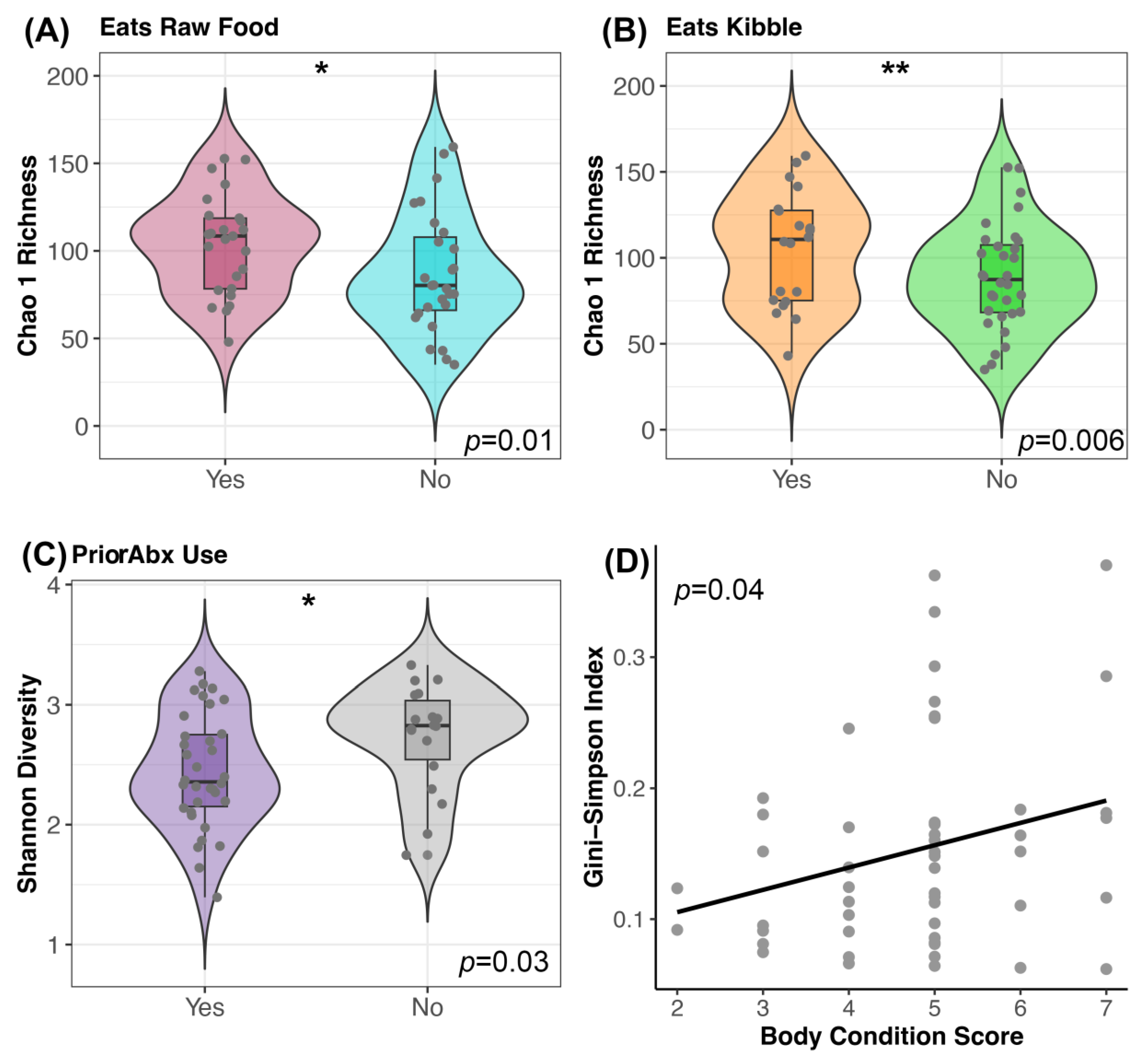
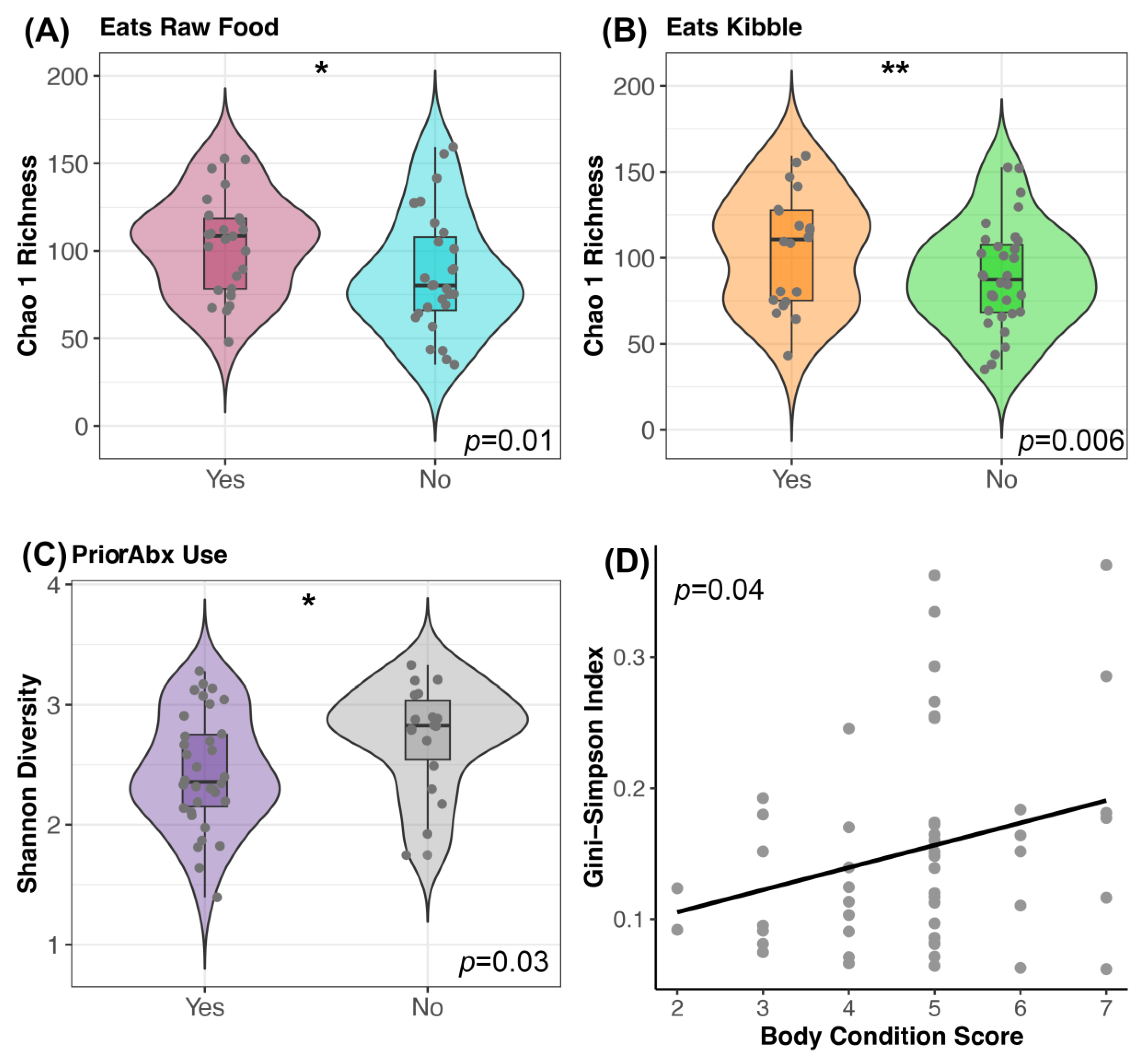
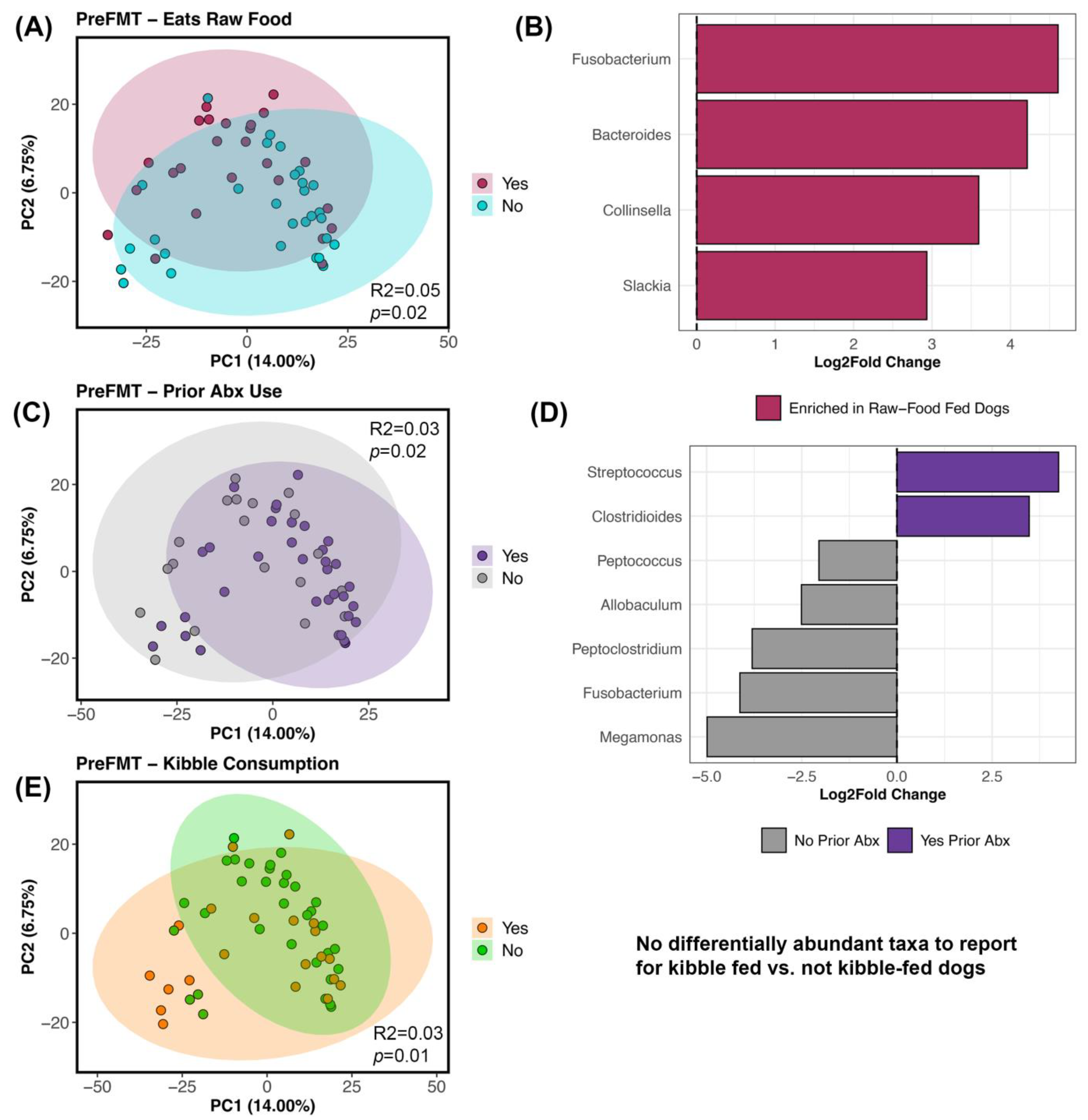
3.3. Host Factors Associated with Microbiome Alpha- and Beta-Diversity
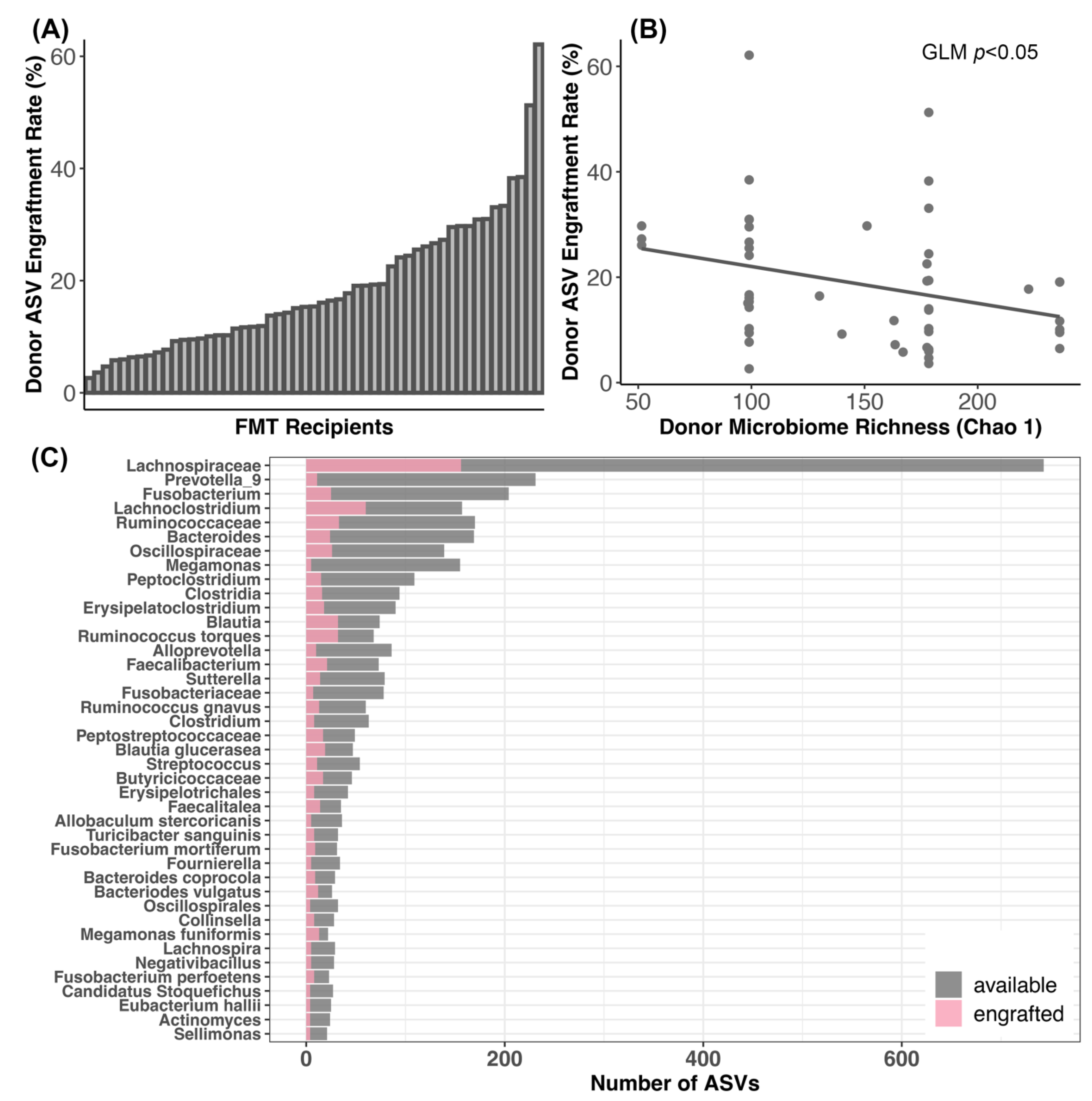
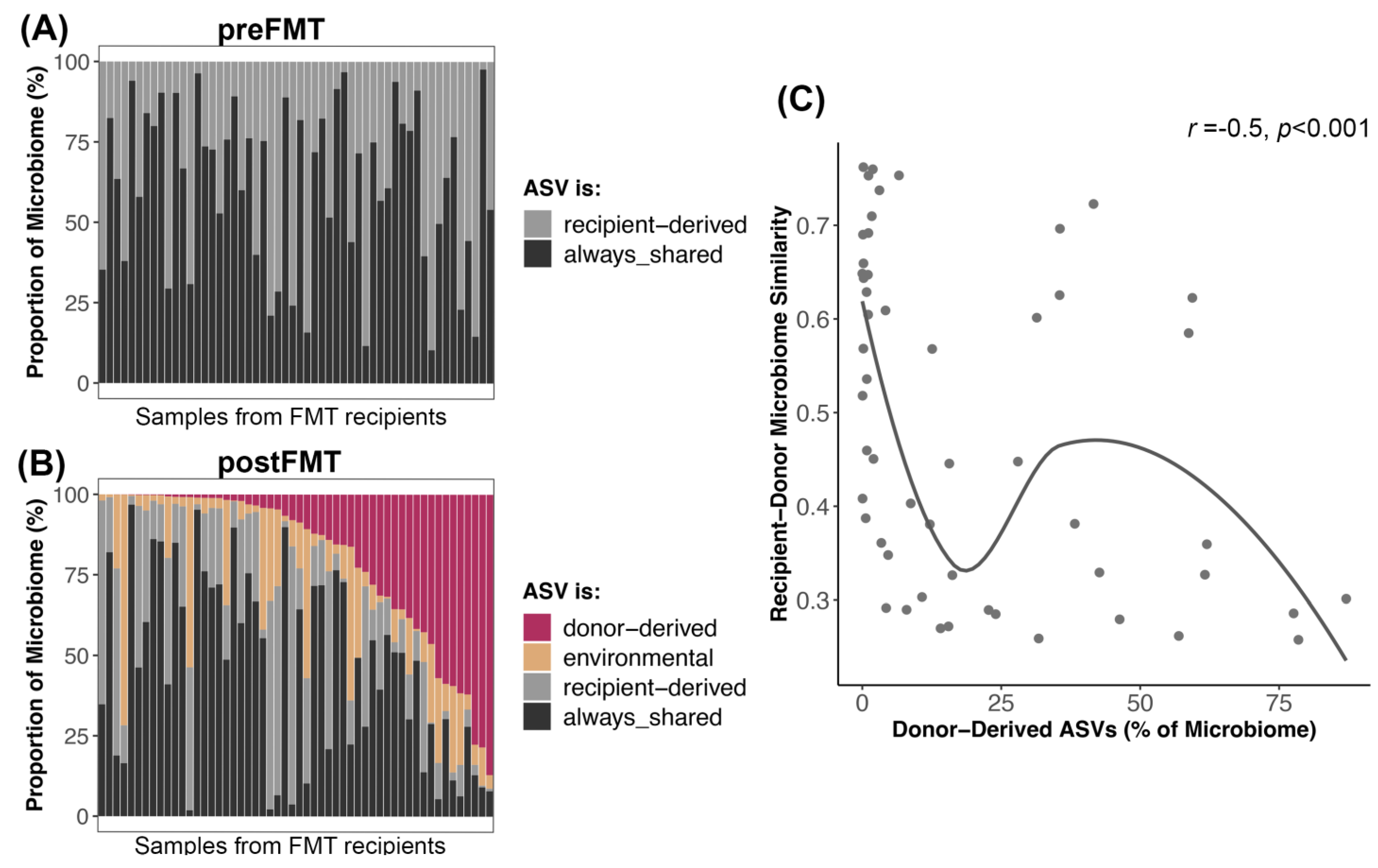
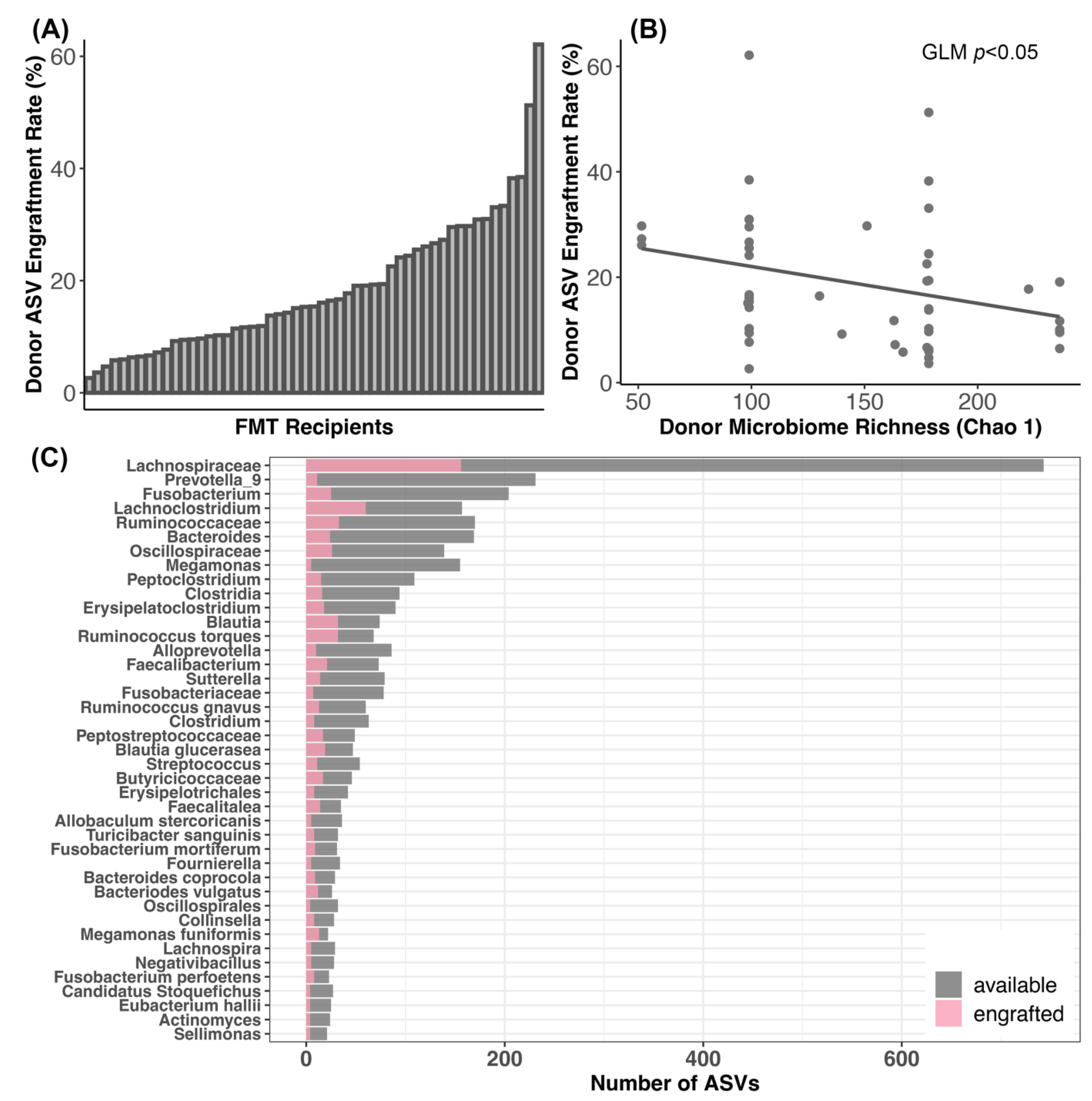
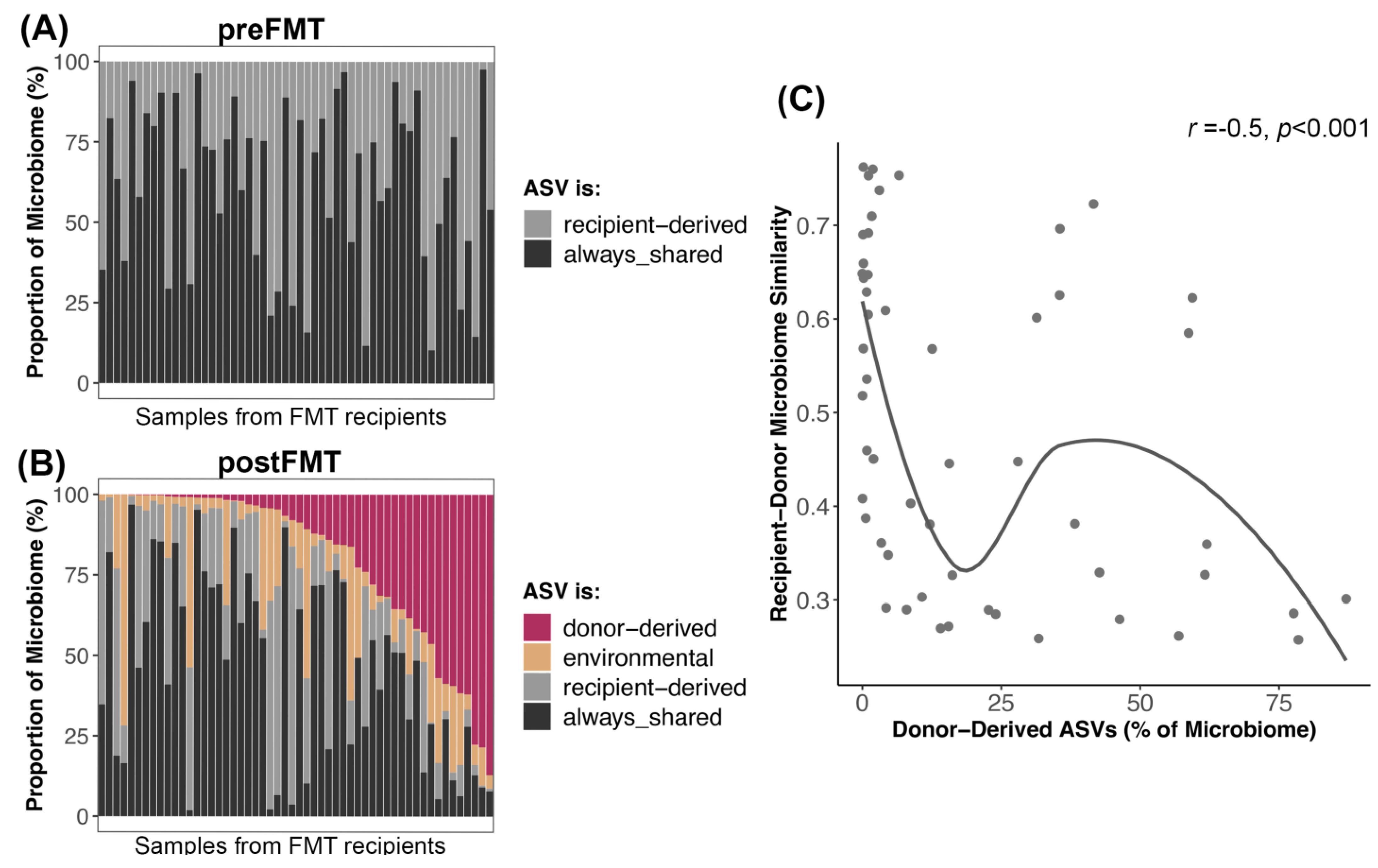
3.4. Bacterial Engraftment after FMT
4. Discussion
4.1. Changes in Canine Fecal Microbiomes after FMT
4.2. Microbiome Associations with Host Factors
4.3. Dynamics of Bacterial Engraftment in Oral FMTs
5. Limitations and Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dandrieux, J.R.S. Inflammatory Bowel Disease versus Chronic Enteropathy in Dogs: Are They One and the Same? J. Small Anim. Pract. 2016, 57, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Kathrani, A. Dietary and Nutritional Approaches to the Management of Chronic Enteropathy in Dogs and Cats. Vet. Clin. N. Am. Small Anim. Pract. 2021, 51, 123–136. [Google Scholar] [CrossRef]
- Segarra, S.; Martínez-Subiela, S.; Cerdà-Cuéllar, M.; Martínez-Puig, D.; Muñoz-Prieto, A.; Rodríguez-Franco, F.; Rodríguez-Bertos, A.; Allenspach, K.; Velasco, A.; Cerón, J. Oral Chondroitin Sulfate and Prebiotics for the Treatment of Canine Inflammatory Bowel Disease: A Randomized, Controlled Clinical Trial. BMC Vet. Res. 2016, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Isidori, M.; Corbee, R.J.; Trabalza-Marinucci, M. Nonpharmacological Treatment Strategies for the Management of Canine Chronic Inflammatory Enteropathy—A Narrative Review. Vet. Sci. 2022, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Cerquetella, M.; Gavazza, A.; Galosi, L.; Berardi, S.; Mangiaterra, S.; Mari, S.; Suchodolski, J.S.; Lidbury, J.A.; Steiner, J.M.; et al. Rapid Resolution of Large Bowel Diarrhea after the Administration of a Combination of a High-Fiber Diet and a Probiotic Mixture in 30 Dogs. Vet. Sci. China 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Dandrieux, J.; Martinez Lopez, L.M.; Prakash, N.; Mansfield, C.S. Treatment Response and Long Term Follow up in Nineteen Dogs Diagnosed with Chronic Enteropathy in Australia. Aust. Vet. J. 2019, 97, 301–307. [Google Scholar] [CrossRef]
- Menozzi, A.; Dall’Aglio, M.; Quintavalla, F.; Dallavalle, L.; Meucci, V.; Bertini, S. Rifaximin is an Effective Alternative to Metronidazole for the Treatment of Chronic Enteropathy in Dogs: A Randomised Trial. BMC Vet. Res. 2016, 12, 217. [Google Scholar] [CrossRef]
- Guard, B.C.; Honneffer, J.B.; Jergens, A.E.; Jonika, M.M.; Toresson, L.; Lawrence, Y.A.; Webb, C.B.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; et al. Longitudinal Assessment of Microbial Dysbiosis, Fecal Unconjugated Bile Acid Concentrations, and Disease Activity in Dogs with Steroid-Responsive Chronic Inflammatory Enteropathy. J. Vet. Intern. Med. 2019, 33, 1295–1305. [Google Scholar] [CrossRef]
- Dandrieux, J.R.S.; Noble, P.-J.M.; Scase, T.J.; Cripps, P.J.; German, A.J. Comparison of a Chlorambucil-Prednisolone Combination with an Azathioprine-Prednisolone Combination for Treatment of Chronic Enteropathy with Concurrent Protein-Losing Enteropathy in Dogs: 27 Cases (2007–2010). J. Am. Vet. Med. Assoc. 2013, 242, 1705–1714. [Google Scholar] [CrossRef]
- Heilmann, R.M.; Steiner, J.M. Clinical Utility of Currently Available Biomarkers in Inflammatory Enteropathies of Dogs. J. Vet. Intern. Med. 2018, 32, 1495–1508. [Google Scholar] [CrossRef]
- Dandrieux, J.R.S.; Mansfield, C.S. Chronic Enteropathy in Canines: Prevalence, Impact and Management Strategies. Vet. Med. Res. Rep. 2019, 10, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Chaitman, J.; Gaschen, F. Fecal Microbiota Transplantation in Dogs. Vet. Clin. N. Am. Small Anim. Pract. 2021, 51, 219–233. [Google Scholar] [CrossRef]
- Brandt, L.J.; Aroniadis, O.C. An Overview of Fecal Microbiota Transplantation: Techniques, Indications, and Outcomes. Gastrointest. Endosc. 2013, 78, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Barko, P.C.; Biggs, P.J.; Gedye, K.R.; Midwinter, A.C.; Williams, D.A.; Burchell, R.K.; Pazzi, P. One Dog’s Waste is Another Dog’s Wealth: A Pilot Study of Fecal Microbiota Transplantation in Dogs with Acute Hemorrhagic Diarrhea Syndrome. PLoS ONE 2021, 16, e0250344. [Google Scholar] [CrossRef] [PubMed]
- Jugan, M.C.; KuKanich, K.; Freilich, L. Clinical Response in Dogs with Acute Hemorrhagic Diarrhea Syndrome Following Randomized Probiotic Treatment or Fecal Microbiota Transplant. Front. Vet. Sci. 2023, 10, 1050538. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.Q.; Gomes, L.A.; Santos, I.S.; Alfieri, A.F.; Weese, J.S.; Costa, M.C. Fecal Microbiota Transplantation in Puppies with Canine Parvovirus Infection. J. Vet. Intern. Med. 2018, 32, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Shima, A.; Takahashi, K.; Matsuda, Y.; Miyajima, M.; Hirokawa, M.; Kondo, H.; Kimura, J.; Ishihara, G.; Ohmori, K. Successful Outcome after a Single Endoscopic Fecal Microbiota Transplantation in a Shiba Dog with Non-Responsive Enteropathy during the Treatment with Chlorambucil. J. Vet. Med. Sci. 2021, 83, 984–989. [Google Scholar] [CrossRef]
- Berlanda, M.; Innocente, G.; Simionati, B.; Di Camillo, B.; Facchin, S.; Giron, M.C.; Savarino, E.; Sebastiani, F.; Fiorio, F.; Patuzzi, I. Faecal Microbiome Transplantation as a Solution to Chronic Enteropathies in Dogs: A Case Study of Beneficial Microbial Evolution. Animals 2021, 11, 1433. [Google Scholar] [CrossRef]
- Bottero, E.; Benvenuti, E.; Ruggiero, P. Fecal Microbiota Transplantation (FMT) in 16 Dogs with Idiopatic IBD. Veterinaria 2017, 31, 31–45. [Google Scholar]
- Niina, A.; Kibe, R.; Suzuki, R.; Yuchi, Y.; Teshima, T.; Matsumoto, H.; Kataoka, Y.; Koyama, H. Improvement in Clinical Symptoms and Fecal Microbiome after Fecal Microbiota Transplantation in a Dog with Inflammatory Bowel Disease. Vet. Med. Res. Rep. 2019, 10, 197–201. [Google Scholar] [CrossRef]
- Niina, A.; Kibe, R.; Suzuki, R.; Yuchi, Y.; Teshima, T.; Matsumoto, H.; Kataoka, Y.; Koyama, H. Fecal Microbiota Transplantation as a New Treatment for Canine Inflammatory Bowel Disease. Biosci. Microbiota Food Health 2021, 40, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Toresson, L.; Spillmann, T.; Pilla, R.; Ludvigsson, U.; Hellgren, J.; Olmedal, G.; Suchodolski, J.S. Clinical Effects of Faecal Microbiota Transplantation as Adjunctive Therapy in Dogs with Chronic Enteropathies—A Retrospective Case Series of 41 Dogs. Vet. Sci. 2023, 10, 271. [Google Scholar] [CrossRef]
- Innocente, G.; Patuzzi, I.; Furlanello, T.; Di Camillo, B.; Bargelloni, L.; Giron, M.C.; Facchin, S.; Savarino, E.; Azzolin, M.; Simionati, B. Machine Learning and Canine Chronic Enteropathies: A New Approach to Investigate FMT Effects. Vet. Sci. China 2022, 9, 502. [Google Scholar] [CrossRef] [PubMed]
- Kerem, U. Fecal Microbiota Transplantation Capsule Therapy via Oral Route for Combatting Atopic Dermatitis in Dogs. Ank. Üniv. Vet. Fak. Dergisi 2022, 69, 211–219. [Google Scholar]
- Sugita, K.; Shima, A.; Takahashi, K.; Ishihara, G.; Kawano, K.; Ohmori, K. Pilot Evaluation of a Single Oral Fecal Microbiota Transplantation for Canine Atopic Dermatitis. Sci. Rep. 2023, 13, 8824. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.A.; Entrolezo, Z.; Jarett, J.K.; Jospin, G.; Kingsbury, D.D.; Martin, A.; Eisen, J.A.; Ganz, H.H. Microbiome Responses to Fecal Microbiota Transplantation in Cats with Chronic Digestive Issues. Vet. Sci. China 2023, 10, 561. [Google Scholar] [CrossRef]
- Smillie, C.S.; Sauk, J.; Gevers, D.; Friedman, J.; Sung, J.; Youngster, I.; Hohmann, E.L.; Staley, C.; Khoruts, A.; Sadowsky, M.J.; et al. Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 2018, 23, 229–240.e5. [Google Scholar] [CrossRef]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.-S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA Gene Sequencing and Analysis Protocol for the Illumina MiniSeq Platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2013, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; He, K.; Chen, J.; Zhang, X. LinDA: Linear Models for Differential Abundance Analysis of Microbiome Compositional Data. Genome Biol. 2022, 23, 95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Zhou, H. GUniFrac, Version 1.7. Generalized UniFrac Distances, Distance-Based Multivariate Methods and Feature-Based Univariate Methods for Microbiome Data Analysis. R Package: Vienna, Austria, 2023.
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Lenth, R.V. Emmeans, Version 1.8.7. Estimated Marginal Means, Aka Least-Squares Means. R Package: Vienna, Austria, 2023.
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef]
- Martinez Arbizu, P. pairwiseAdonis: Pairwise Multilevel Comparison Using Adonis 2017; R Package: Vienna, Austria, 2019. [Google Scholar]
- Gilbert, J.A.; Lynch, S.V. Community Ecology as a Framework for Human Microbiome Research. Nat. Med. 2019, 25, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.S. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352–359. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic Analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Carapeto, S.; Cunha, E.; Serrano, I.; Pascoal, P.; Pereira, M.; Abreu, R.; Neto, S.; Antunes, B.; Dias, R.; Tavares, L.; et al. Effect of the Administration of a Lyophilised Faecal Capsules on the Intestinal Microbiome of Dogs: A Pilot Study. Genes 2023, 14, 1676. [Google Scholar] [CrossRef]
- Suchodolski, J.S.; Markel, M.E.; Garcia-Mazcorro, J.F.; Unterer, S.; Heilmann, R.M.; Dowd, S.E.; Kachroo, P.; Ivanov, I.; Minamoto, Y.; Dillman, E.M.; et al. The Fecal Microbiome in Dogs with Acute Diarrhea and Idiopathic Inflammatory Bowel Disease. PLoS ONE 2012, 7, e51907. [Google Scholar] [CrossRef]
- Pilla, R.; Guard, B.C.; Blake, A.B.; Ackermann, M.; Webb, C.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Long-Term Recovery of the Fecal Microbiome and Metabolome of Dogs with Steroid-Responsive Enteropathy. Animals 2021, 11, 2498. [Google Scholar] [CrossRef] [PubMed]
- Ziese, A.-L.; Suchodolski, J.S.; Hartmann, K.; Busch, K.; Anderson, A.; Sarwar, F.; Sindern, N.; Unterer, S. Effect of Probiotic Treatment on the Clinical Course, Intestinal Microbiome, and Toxigenic Clostridium Perfringens in Dogs with Acute Hemorrhagic Diarrhea. PLoS ONE 2018, 13, e0204691. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, F.; Hou, Q.; Huang, W.; Liu, Y.; Zhang, H.; Sun, Z. Metagenomic Analysis Revealed Beneficial Effects of Probiotics in Improving the Composition and Function of the Gut Microbiota in Dogs with Diarrhoea. Food Funct. 2019, 10, 2618–2629. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.J. Role of Colonic Short-Chain Fatty Acid Transport in Diarrhea. Annu. Rev. Physiol. 2010, 72, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.N. Energy Contributions of Volatile Fatty Acids from the Gastrointestinal Tract in Various Species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef]
- Kamath, P.S.; Hoepfner, M.T.; Phillips, S.F. Short-Chain Fatty Acids Stimulate Motility of the Canine Ileum. Am. J. Physiol. 1987, 253, G427–G433. [Google Scholar] [CrossRef]
- Sandri, M.; Dal Monego, S.; Conte, G.; Sgorlon, S.; Stefanon, B. Raw Meat Based Diet Influences Faecal Microbiome and End Products of Fermentation in Healthy Dogs. BMC Vet. Res. 2017, 13, 65. [Google Scholar] [CrossRef]
- Chaitman, J.; Ziese, A.-L.; Pilla, R.; Minamoto, Y.; Blake, A.B.; Guard, B.C.; Isaiah, A.; Lidbury, J.A.; Steiner, J.M.; Unterer, S.; et al. Fecal Microbial and Metabolic Profiles in Dogs with Acute Diarrhea Receiving Either Fecal Microbiota Transplantation or Oral Metronidazole. Front. Vet. Sci. 2020, 7, 192. [Google Scholar] [CrossRef]
- Beloshapka, A.N.; Dowd, S.E.; Suchodolski, J.S.; Steiner, J.M.; Duclos, L.; Swanson, K.S. Fecal Microbial Communities of Healthy Adult Dogs Fed Raw Meat-Based Diets with or without Inulin or Yeast Cell Wall Extracts as Assessed by 454 Pyrosequencing. FEMS Microbiol. Ecol. 2013, 84, 532–541. [Google Scholar] [CrossRef]
- Bermingham, E.N.; Maclean, P.; Thomas, D.G.; Cave, N.J.; Young, W. Key Bacterial Families (Clostridiaceae, Erysipelotrichaceae and Bacteroidaceae) Are Related to the Digestion of Protein and Energy in Dogs. PeerJ 2017, 5, e3019. [Google Scholar] [CrossRef]
- Moinard, A.; Payen, C.; Ouguerram, K.; André, A.; Hernandez, J.; Drut, A.; Biourge, V.C.; Suchodolski, J.S.; Flanagan, J.; Nguyen, P.; et al. Effects of High-Fat Diet at Two Energetic Levels on Fecal Microbiota, Colonic Barrier, and Metabolic Parameters in Dogs. Front. Vet. Sci. 2020, 7, 566282. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, S.; Ariza, G.; Rincón-Riveros, A.; Muñoz, M.; Ramírez, J.D. Diet-Induced Changes in Fecal Microbiota Composition and Diversity in Dogs (Canis Lupus Familiaris): A Comparative Study of BARF-Type and Commercial Diets. Comp. Immunol. Microbiol. Infect. Dis. 2023, 98, 102007. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Becker, A.A.M.J.; Luo, Y.; Zhang, W.; Ge, B.; Leng, C.; Wang, G.; Ding, L.; Wang, J.; Fu, X.; et al. The Fecal Microbiota of Dogs Switching to a Raw Diet Only Partially Converges to That of Wolves. Front. Microbiol. 2021, 12, 701439. [Google Scholar] [CrossRef] [PubMed]
- Pilla, R.; Gaschen, F.P.; Barr, J.W.; Olson, E.; Honneffer, J.; Guard, B.C.; Blake, A.B.; Villanueva, D.; Khattab, M.R.; AlShawaqfeh, M.K.; et al. Effects of Metronidazole on the Fecal Microbiome and Metabolome in Healthy Dogs. J. Vet. Intern. Med. 2020, 34, 1853–1866. [Google Scholar] [CrossRef] [PubMed]
- Stavroulaki, E.M.; Suchodolski, J.S.; Pilla, R.; Fosgate, G.T.; Sung, C.-H.; Lidbury, J.A.; Steiner, J.M.; Xenoulis, P.G. Short- and Long-Term Effects of Amoxicillin/Clavulanic Acid or Doxycycline on the Gastrointestinal Microbiome of Growing Cats. PLoS ONE 2021, 16, e0253031. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Dowd, S.E.; Westermarck, E.; Steiner, J.M.; Wolcott, R.D.; Spillmann, T.; Harmoinen, J.A. The Effect of the Macrolide Antibiotic Tylosin on Microbial Diversity in the Canine Small Intestine as Demonstrated by Massive Parallel 16S rRNA Gene Sequencing. BMC Microbiol. 2009, 9, 210. [Google Scholar] [CrossRef]
- Reygaert, W.C. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Antibiotic Resistance Mechanisms of Clinically Important Bacteria. Medicina 2011, 47, 137–146. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Metcalf, B.J.; Chochua, S.; Gertz, R.E.; Hawkins, P.A.; Ricaldi, J.; Li, Z.; Walker, H.; Tran, T.; Rivers, J.; Mathis, S.; et al. Short-Read Whole Genome Sequencing for Determination of Antimicrobial Resistance Mechanisms and Capsular Serotypes of Current Invasive Streptococcus Agalactiae Recovered in the USA. Clin. Microbiol. Infect. 2017, 23, 574.e7–574.e14. [Google Scholar] [CrossRef]
- Kimura, K.; Suzuki, S.; Wachino, J.-I.; Kurokawa, H.; Yamane, K.; Shibata, N.; Nagano, N.; Kato, H.; Shibayama, K.; Arakawa, Y. First Molecular Characterization of Group B Streptococci with Reduced Penicillin Susceptibility. Antimicrob. Agents Chemother. 2008, 52, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.M.; Lynch, T.; McCorrister, S.; Kibsey, P.; Miller, M.; Gravel, D.; Westmacott, G.R.; Mulvey, M.R. Canadian Nosocomial Infection Surveillance Program (CNISP) Proteomic Analysis of a NAP1 Clostridium Difficile Clinical Isolate Resistant to Metronidazole. PLoS ONE 2014, 9, e82622. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Chong, P.; Zhang, J.; Hizon, R.; Du, T.; Graham, M.R.; Beniac, D.R.; Booth, T.F.; Kibsey, P.; Miller, M.; et al. Characterization of a Stable, Metronidazole-Resistant Clostridium Difficile Clinical Isolate. PLoS ONE 2013, 8, e53757. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.L.; Ji, S.Y.; Lee, S.D.; Lee, Y.K.; Kim, B.; Kim, K.H. Difference of Gut Microbiota Composition Based on the Body Condition Scores in Dogs. J. Anim. Sci. Technol. 2020, 62, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lauber, C.L.; Czarnecki-Maulden, G.; Pan, Y.; Hannah, S.S. Effects of the Dietary Protein and Carbohydrate Ratio on Gut Microbiomes in Dogs of Different Body Conditions. mBio 2017, 8, e01703-16. [Google Scholar] [CrossRef]
- Thomson, P.; Santibáñez, R.; Rodríguez-Salas, C.; Flores-Yañez, C.; Garrido, D. Differences in the Composition and Predicted Functions of the Intestinal Microbiome of Obese and Normal Weight Adult Dogs. PeerJ 2022, 10, e12695. [Google Scholar] [CrossRef]
- Singh, P.; Alm, E.J.; Kelley, J.M.; Cheng, V.; Smith, M.; Kassam, Z.; Nee, J.; Iturrino, J.; Lembo, A. Effect of Antibiotic Pretreatment on Bacterial Engraftment after Fecal Microbiota Transplant (FMT) in IBS-D. Gut Microbes 2022, 14, 2020067. [Google Scholar] [CrossRef]
- Podlesny, D.; Durdevic, M.; Paramsothy, S.; Kaakoush, N.O.; Högenauer, C.; Gorkiewicz, G.; Walter, J.; Fricke, W.F. Identification of Clinical and Ecological Determinants of Strain Engraftment after Fecal Microbiota Transplantation Using Metagenomics. Cell Rep. Med. 2022, 3, 100711. [Google Scholar] [CrossRef]
- Debray, R.; Herbert, R.A.; Jaffe, A.L.; Crits-Christoph, A.; Power, M.E.; Koskella, B. Priority Effects in Microbiome Assembly. Nat. Rev. Microbiol. 2022, 20, 109–121. [Google Scholar] [CrossRef]
- Ianiro, G.; Punčochář, M.; Karcher, N.; Porcari, S.; Armanini, F.; Asnicar, F.; Beghini, F.; Blanco-Míguez, A.; Cumbo, F.; Manghi, P.; et al. Variability of Strain Engraftment and Predictability of Microbiome Composition after Fecal Microbiota Transplantation across Different Diseases. Nat. Med. 2022, 28, 1913–1923. [Google Scholar] [CrossRef]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between Diet, Gut Microbiota Composition and Gut Metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the In Vivo Metabolic Potential of Two Human Gut Acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef] [PubMed]
- Markowiak-Kopeć, P.; Śliżewska, K. The Effect of Probiotics on the Production of Short-Chain Fatty Acids by Human Intestinal Microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.; Chang, E.T.; Wang, J.-Y.; Rao, J.N. Polyamines and Gut Mucosal Homeostasis. J. Gastrointest. Dig. Syst. 2012, 2, 25237589. [Google Scholar] [CrossRef]
- Luo, Y.; Xiao, Y.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. The Role of Mucin and Oligosaccharides via Cross-Feeding Activities by Bifidobacterium: A Review. Int. J. Biol. Macromol. 2021, 167, 1329–1337. [Google Scholar] [CrossRef]
- Crost, E.H.; Tailford, L.E.; Monestier, M.; Swarbreck, D.; Henrissat, B.; Crossman, L.C.; Juge, N. The Mucin-Degradation Strategy of Ruminococcus Gnavus: The Importance of Intramolecular Trans-Sialidases. Gut Microbes 2016, 7, 302–312. [Google Scholar] [CrossRef]
- Olivos Caicedo, K.Y.; Fernandez-Materan, F.V.; Hernandez, A.G.; Daniel, S.L.; Alves, J.M.P.; Ridlon, J.M. Complete Genome Sequence of the Archetype Bile Acid 7α-Dehydroxylating Bacterium, Clostridium Scindens VPI12708, Isolated from Human Feces, circa 1980. Microbiol. Resour. Announc. 2023, 12, e0002923. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Specific Subcategory | N (%) |
|---|---|---|
| Age, in years | median & (range) | 5.2 (1–15) |
| Body condition score * | median & (range) | 5 (2–8) |
| Body weight category | <20 lbs | 13 (24%) |
| 20–40 lbs | 9 (17%) | |
| 40–60 lbs | 13 (24%) | |
| >60 lbs | 19 (35%) | |
| Sex | Female | 25 (46%) |
| Male | 29 (54%) | |
| Breed (broad) | Poodle | 8 (15%) |
| Golden Retriever | 4 (7%) | |
| Terrier | 6 (11%) | |
| German Shepherd | 5 (9%) | |
| Other | 31 (58%) | |
| Diet * (not mutually exclusive) | Eat Kibble | 21 (39%) |
| Eat Raw Food | 25 (46%) | |
| Eat Canned Food | 27 (50%) | |
| Spayed or Neutered | Yes | 42 (78%) |
| No | 12 (22%) | |
| Antibiotics * | Yes | 35 (65%) |
| No | 19 (35%) | |
| Initial clinical signs * | Diarrhea | 26 (48%) |
| Vomiting & Diarrhea | 16 (30%) | |
| Vomiting | 7 (13%) | |
| Constipation & Diarrhea | 5 (9%) |
| Alpha-Diversity | Beta-Diversity | |||
|---|---|---|---|---|
| Predictor | preFMT | postFMT | preFMT | postFMT |
| Clinical signs | ||||
| Raw Food consumption | X | X | ||
| Dry Food consumption | X | X | X | |
| Prior antibiotics use | X | X | X | |
| Body condition score | X | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, C.A.; Entrolezo, Z.; Jarett, J.K.; Jospin, G.; Martin, A.; Ganz, H.H. Microbiome Responses to Oral Fecal Microbiota Transplantation in a Cohort of Domestic Dogs. Vet. Sci. 2024, 11, 42. https://doi.org/10.3390/vetsci11010042
Rojas CA, Entrolezo Z, Jarett JK, Jospin G, Martin A, Ganz HH. Microbiome Responses to Oral Fecal Microbiota Transplantation in a Cohort of Domestic Dogs. Veterinary Sciences. 2024; 11(1):42. https://doi.org/10.3390/vetsci11010042
Chicago/Turabian StyleRojas, Connie A., Zhandra Entrolezo, Jessica K. Jarett, Guillaume Jospin, Alex Martin, and Holly H. Ganz. 2024. "Microbiome Responses to Oral Fecal Microbiota Transplantation in a Cohort of Domestic Dogs" Veterinary Sciences 11, no. 1: 42. https://doi.org/10.3390/vetsci11010042
APA StyleRojas, C. A., Entrolezo, Z., Jarett, J. K., Jospin, G., Martin, A., & Ganz, H. H. (2024). Microbiome Responses to Oral Fecal Microbiota Transplantation in a Cohort of Domestic Dogs. Veterinary Sciences, 11(1), 42. https://doi.org/10.3390/vetsci11010042