
A Multifaceted Approach for Evaluating Hepatitis E Virus Infectivity In Vitro: Cell Culture and Innovative Molecular Methods for Integrity Assessment

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses
- (1)
- (2)
- A leftover viremic serum sample from a patient acutely infected with HEV gt3c (National Reference Center for viral hepatitis, Sciensano, Belgium).
2.2. HEV Cell Culture Infection Experiments
2.2.1. Cell Lines and Media
2.2.2. HEV Inoculation
2.2.3. Immunofluorescence Assay
2.3. Capsid Integrity Assays
2.4. RNA Extraction, Purification and Detection
2.5. Genome Integrity Assay: Two-Step Long-Range RT-qPCR
2.6. Statistical Analysis
3. Results
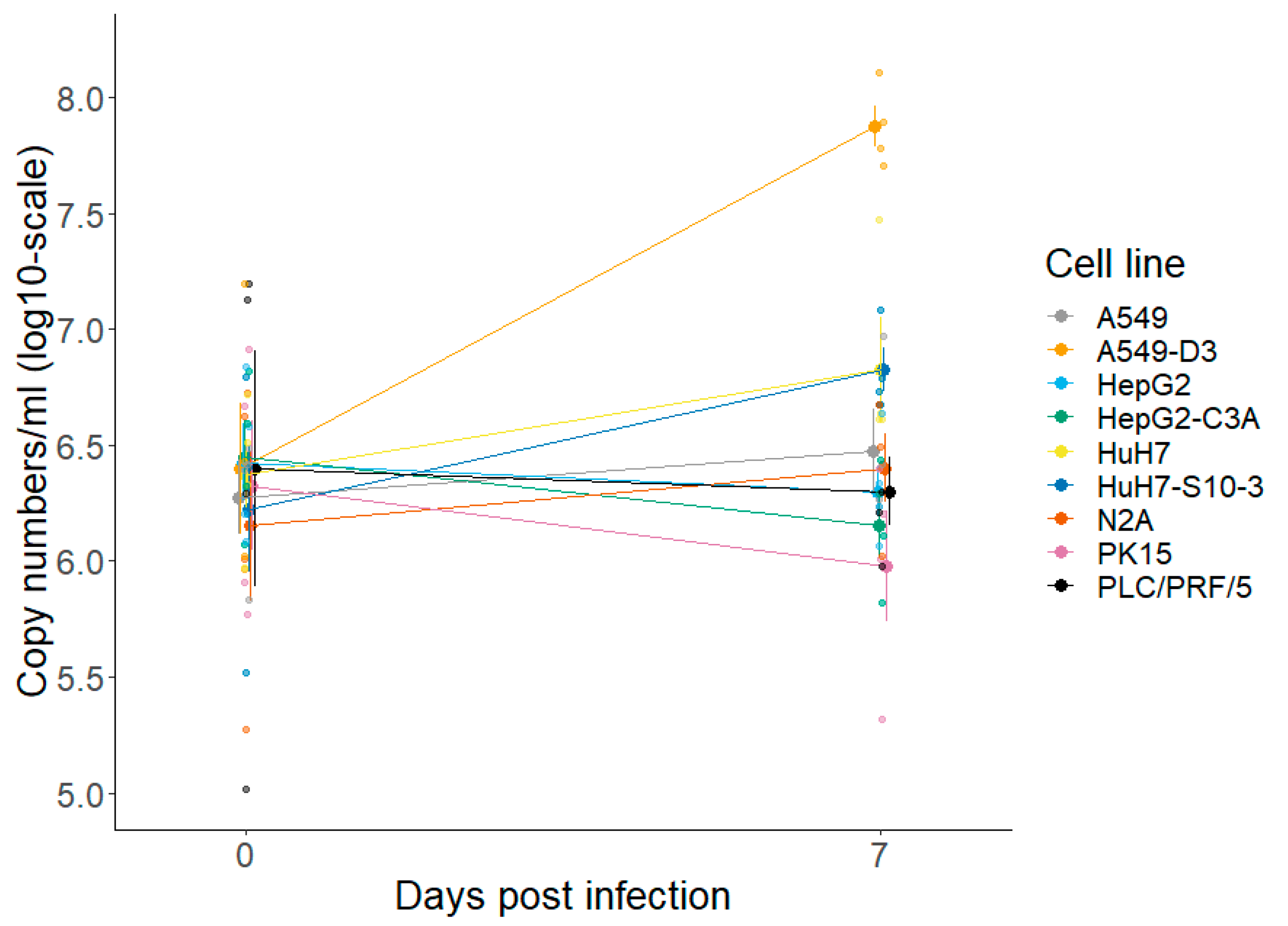
3.1. Evaluation of HEV Cell Culture Models
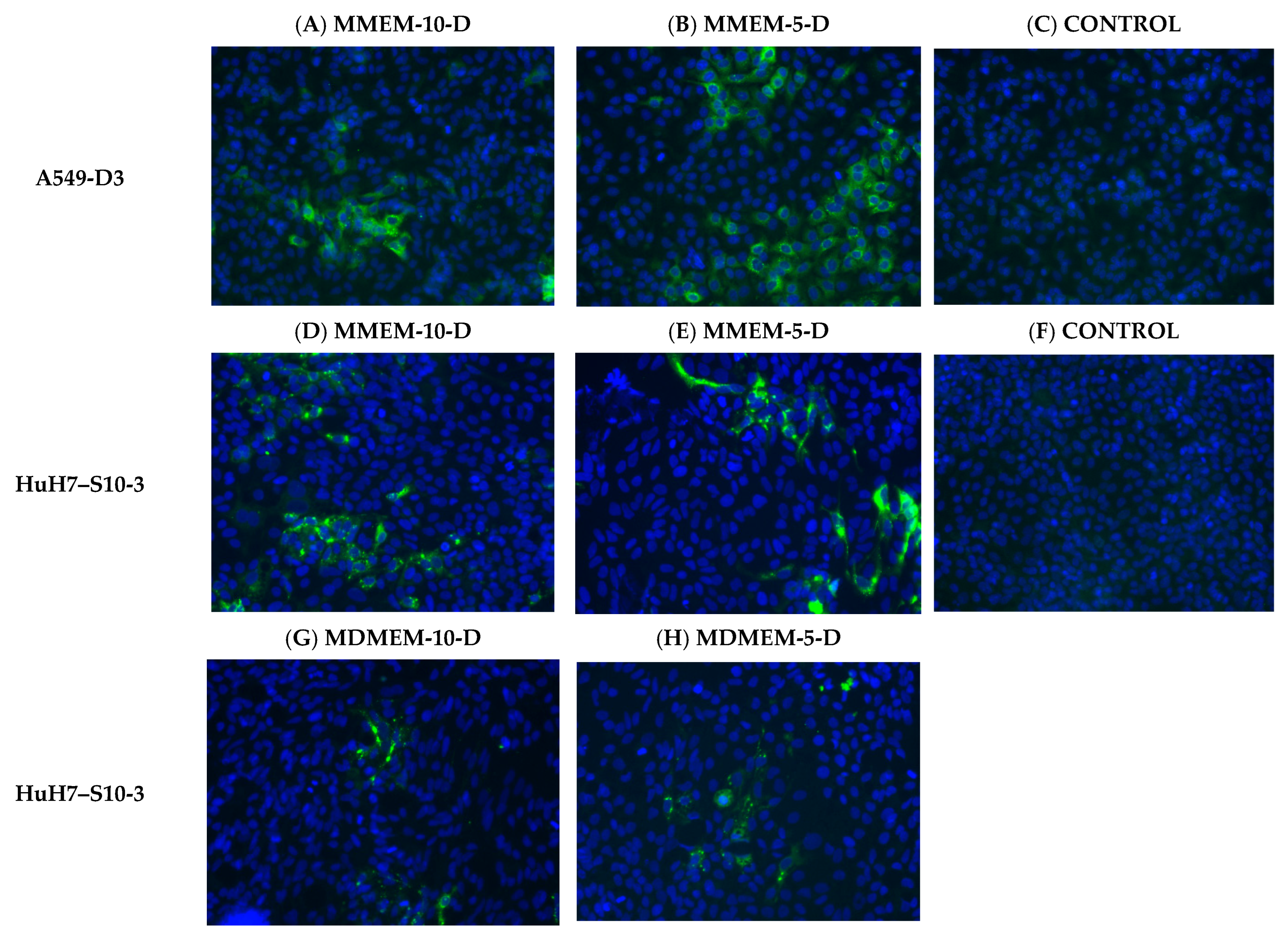
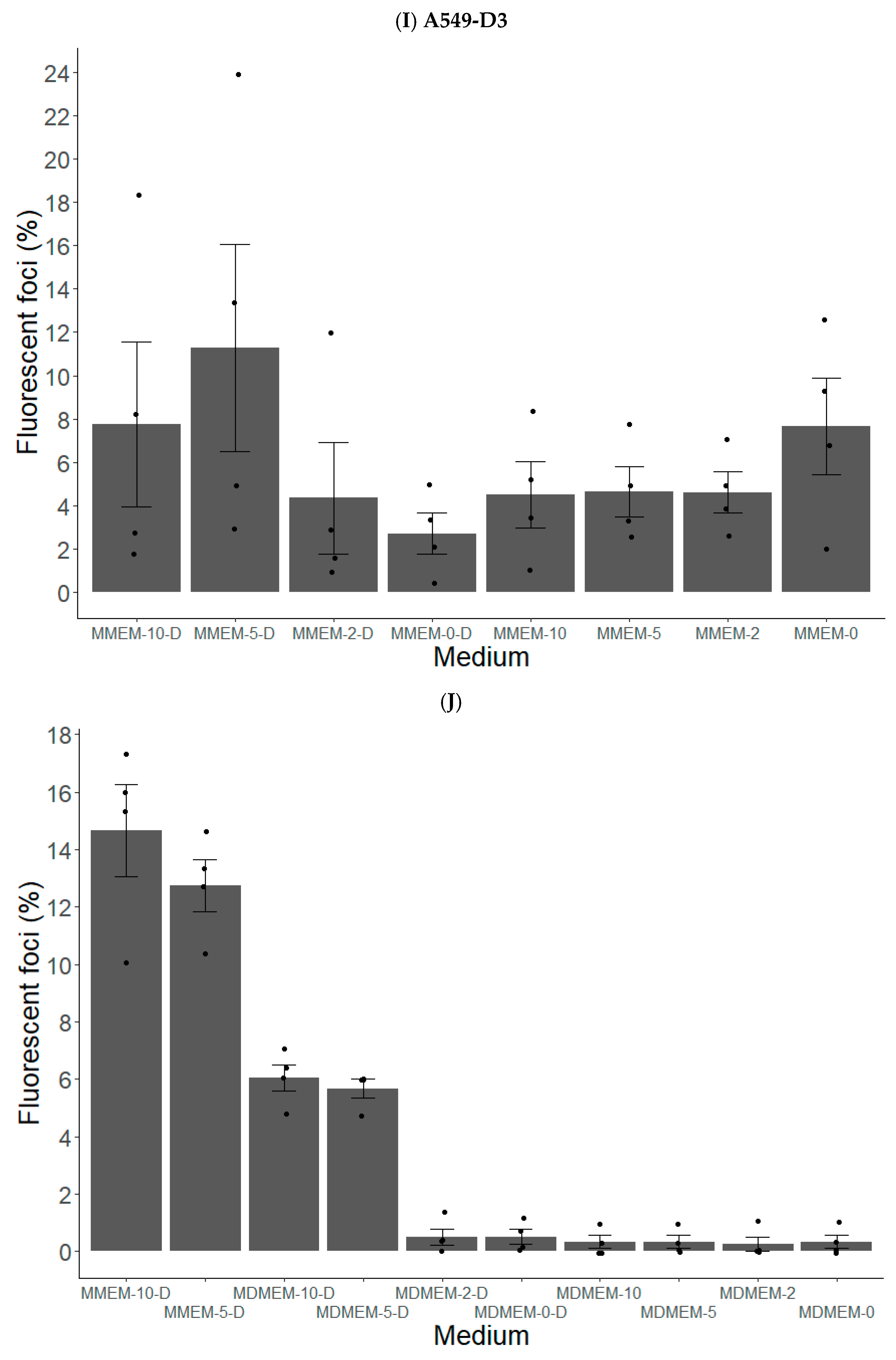
3.1.1. A549-D3 and HuH7-S10-3 Cells Are Promising for a Cell Culture-Based HEV Infection Model
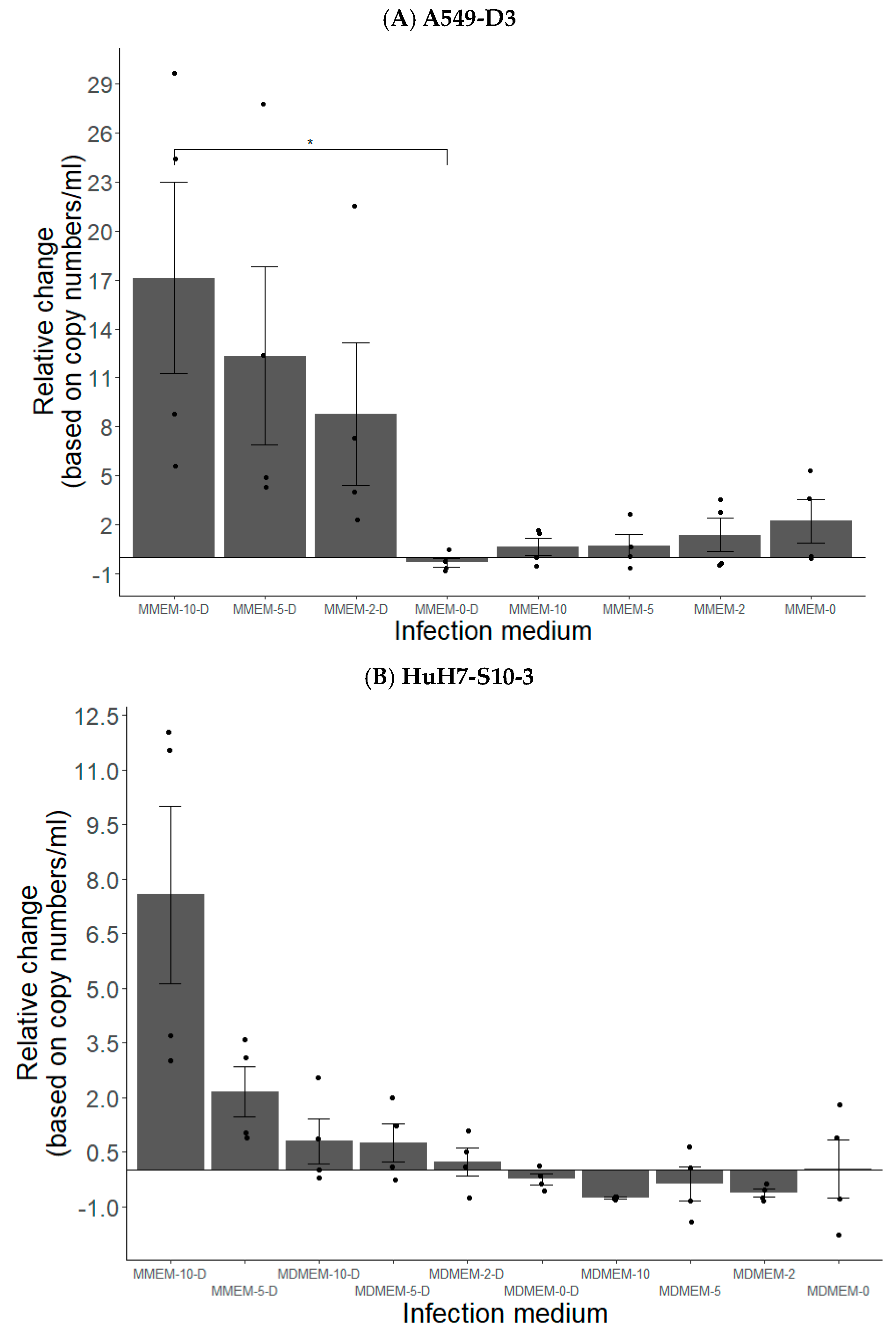
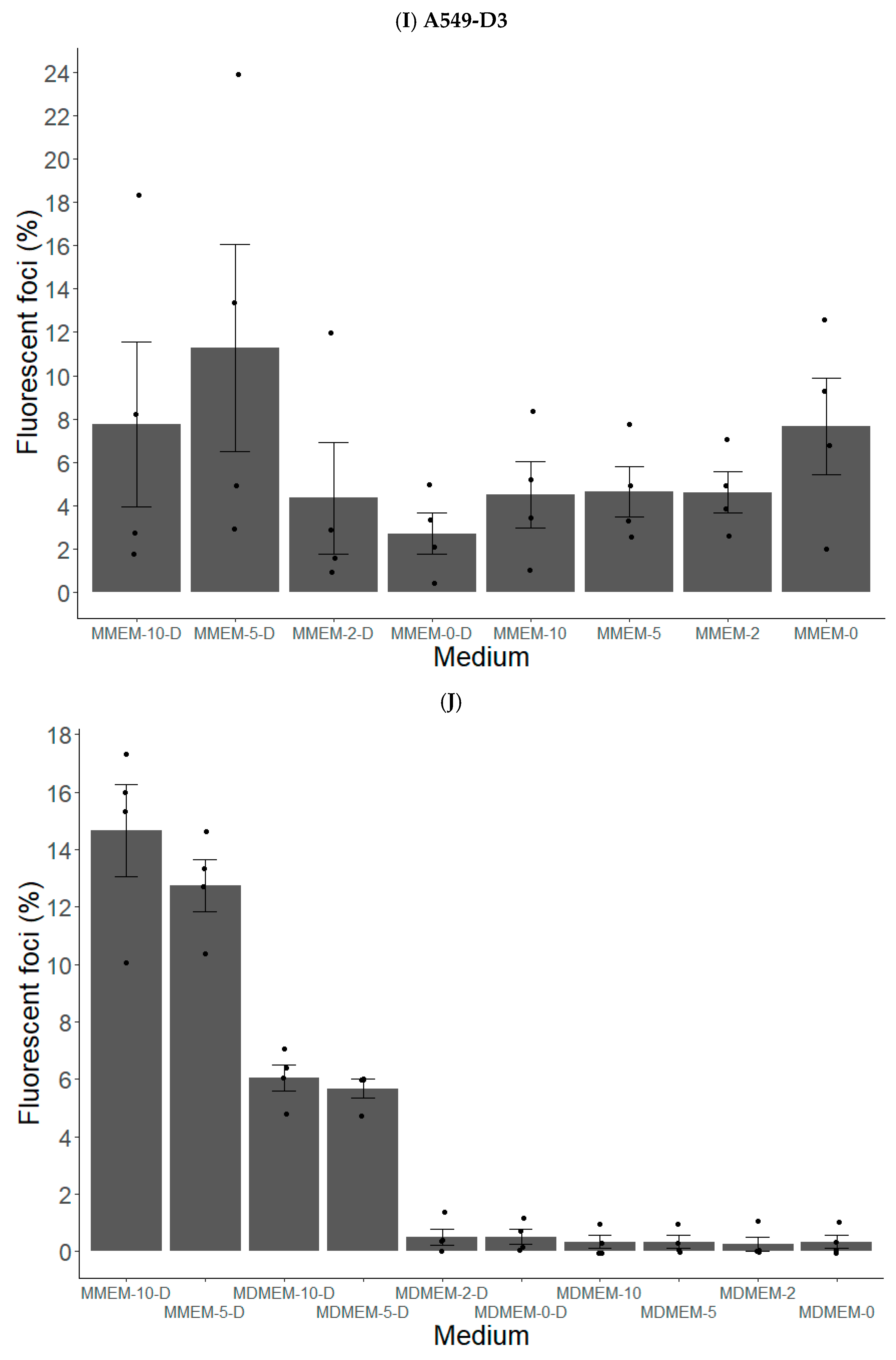
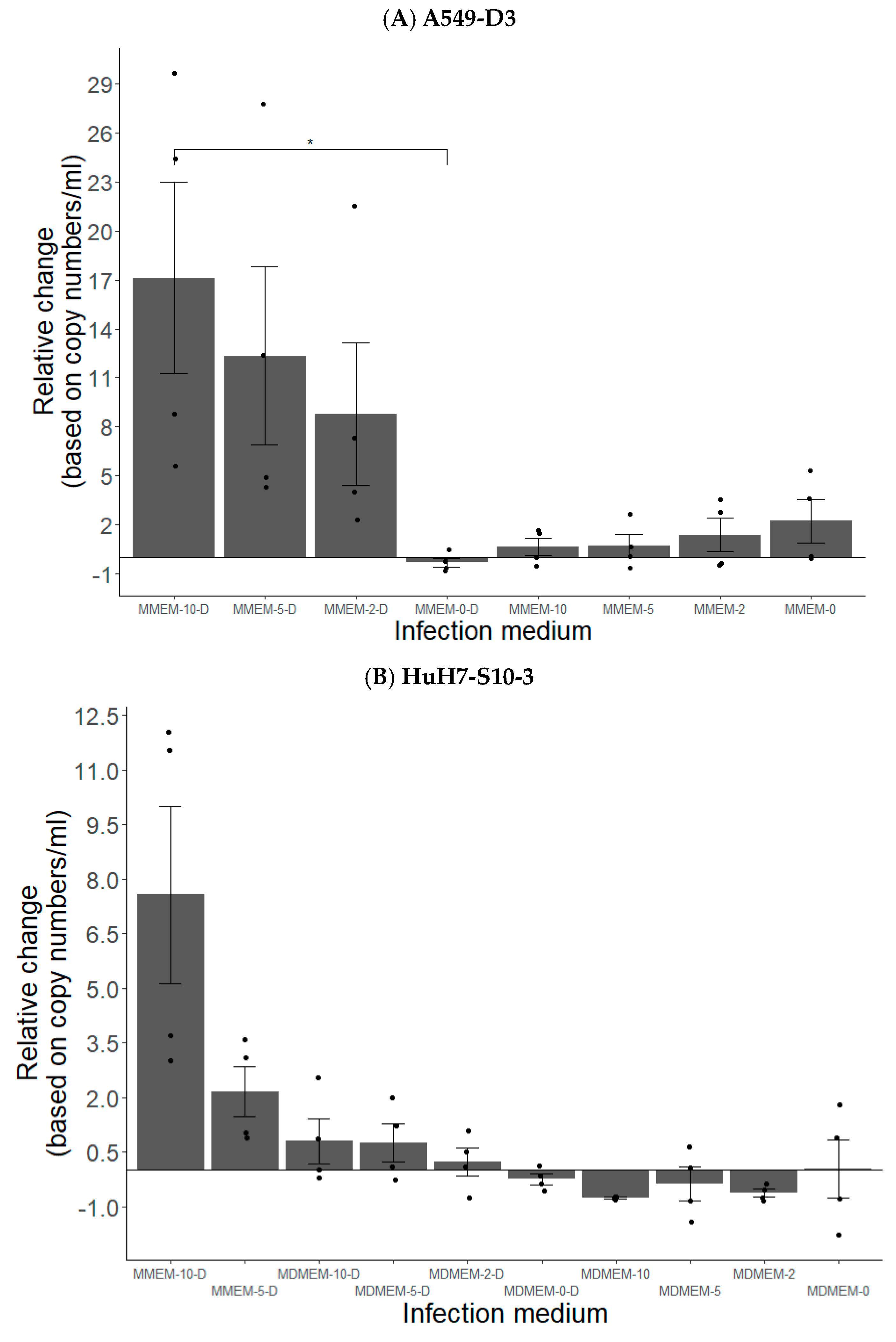
3.1.2. FBS and DMSO Positively Contribute to HEV Propagation in A549-D3 and HuH7-S10-3 Cells
3.1.3. The Peak of HEV Concentration in Supernatant of A549-D3 and HuH7-S10-3 Cells Is Reached after Five and Six Days, Respectively
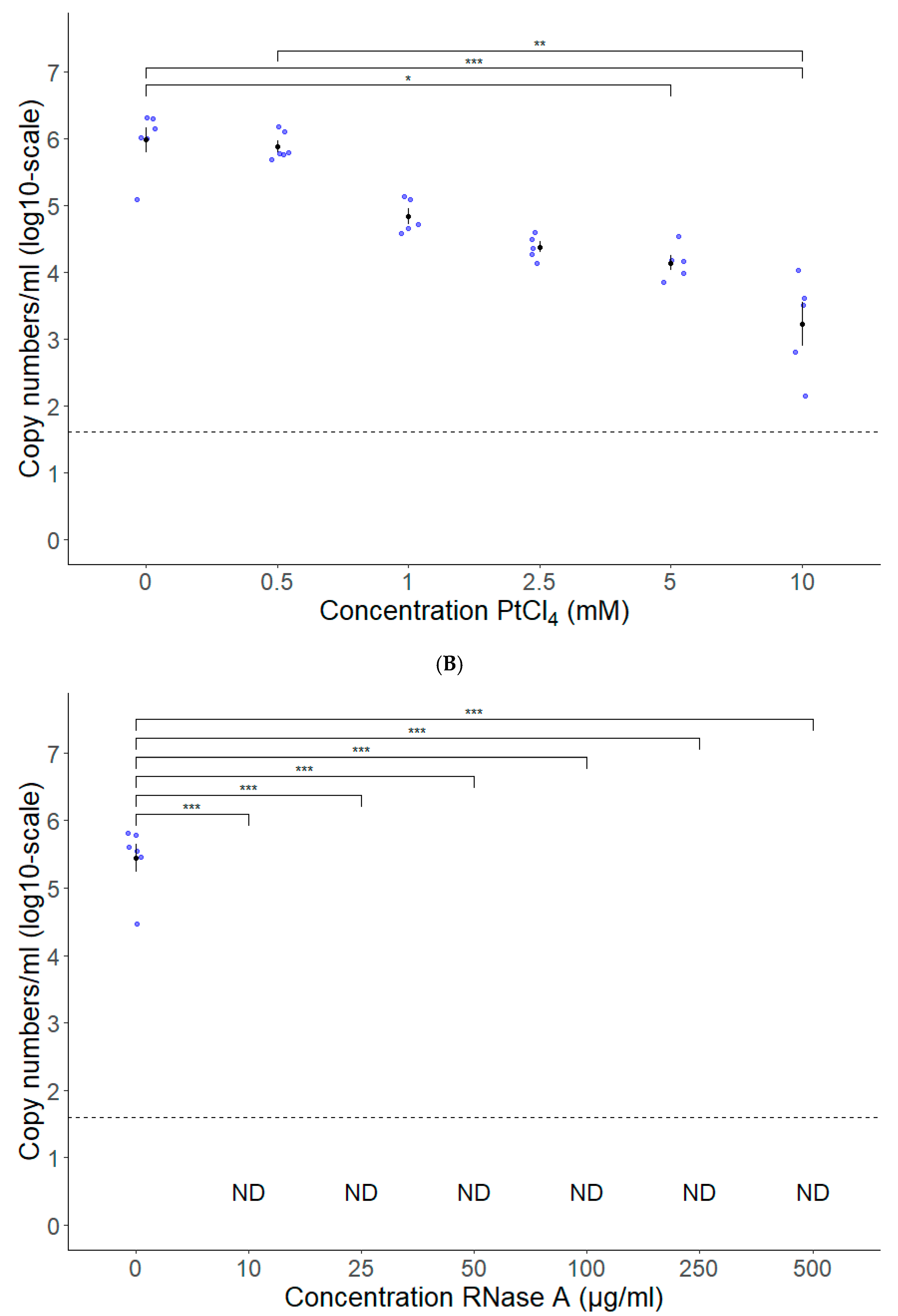
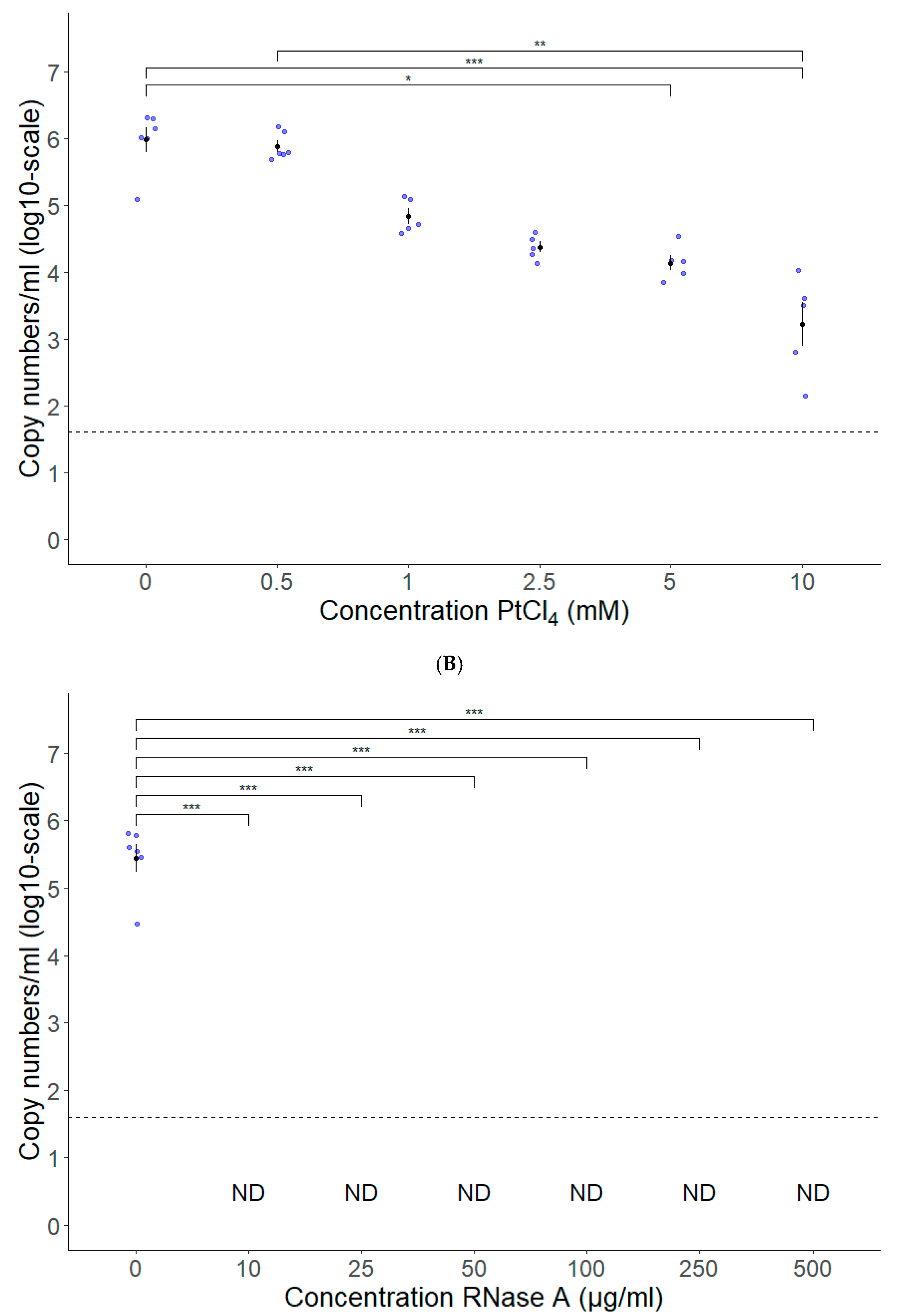
3.2. PtCl4 and RNase A Combined with Conventional RT-qPCR Can Be Used as HEV Capsid Integrity Assays
3.3. Long-Range RT-qPCR Can Be Used as a Proxy for Detecting Intact HEV Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ECDC. Options for National Testing and Surveillance for Hepatitis E Virus in the EU/EEA—Operational Guidance; ECDC: Solna, Sweden, 2019; ISBN 9789294983756. [Google Scholar]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis e Virus: Advances and Challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Schenk, J.; De Somer, T.; Roskams, T.; Locus, T.; Klamer, S.; Subissi, L.; Suin, V.; Delwaide, J.; Stärkel, P.; et al. Viral Clade Is Associated with Severity of Symptomatic Genotype 3 Hepatitis E Virus Infections in Belgium, 2010–2018. J. Hepatol. 2023, 78, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, K.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Monoclonal Antibodies Raised against the ORF3 Protein of Hepatitis e Virus (HEV) Can Capture HEV Particles in Culture Supernatant and Serum but Not Those in Feces. Arch. Virol. 2008, 153, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; Van Der Poel, W.H.M.; Smith, D.B. ICTV Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed Reference Sequences for Subtypes of Hepatitis E Virus (Species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Meng, X.J. Hepatitis E Virus: Host Tropism and Zoonotic Infection. Curr. Opin. Microbiol. 2021, 59, 8–15. [Google Scholar] [CrossRef]
- Lee, G.H.; Tan, B.H.; Chi-Yuan Teo, E.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Kim Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection with Camelid Hepatitis e Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357.e3. [Google Scholar] [CrossRef]
- King, N.J.; Hewitt, J.; Perchec-Merien, A.M. Hiding in Plain Sight? It’s Time to Investigate Other Possible Transmission Routes for Hepatitis E Virus (HEV) in Developed Countries. Food Environ. Virol. 2018, 10, 225–252. [Google Scholar] [CrossRef]
- Sooryanarain, H.; Meng, X.J. Hepatitis E Virus: Reasons for Emergence in Humans. Curr. Opin. Virol. 2019, 34, 10–17. [Google Scholar] [CrossRef]
- Thiry, D.; Mauroy, A.; Saegerman, C.; Licoppe, A.; Fett, T.; Thomas, I.; Brochier, B.; Thiry, E.; Linden, A. Belgian Wildlife as Potential Zoonotic Reservoir of Hepatitis E Virus. Transbound. Emerg. Dis. 2017, 64, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Treagus, S.; Wright, C.; Baker-Austin, C.; Longdon, B.; Lowther, J. The Foodborne Transmission of Hepatitis E Virus to Humans. Food Environ. Virol. 2021, 13, 127–145. [Google Scholar] [CrossRef] [PubMed]
- van der Honing, R.W.H.; van Coillie, E.; Antonis, A.F.G.; van der Poel, W.H.M. First Isolation of Hepatitis E Virus Genotype 4 in Europe through Swine Surveillance in the Netherlands and Belgium. PLoS ONE 2011, 6, e22673. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.A.; Jansen, C.C.C.; Hägele, G.; Zwartkruis-Nahuis, A.; Tijsma, A.S.L.; Vennema, H. Monitoring of Pork Liver and Meat Products on the Dutch Market for the Presence of HEV RNA. Int. J. Food Microbiol. 2019, 296, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Boxman, I.L.A.; Jansen, C.C.C.; Zwartkruis-Nahuis, A.J.T.; Hägele, G.; Sosef, N.P.; Dirks, R.A.M. Detection and Quantification of Hepatitis E Virus RNA in Ready to Eat Raw Pork Sausages in the Netherlands. Int. J. Food Microbiol. 2020, 333, 108791. [Google Scholar] [CrossRef] [PubMed]
- Pallerla, S.R.; Schembecker, S.; Meyer, C.G.; Linh, L.T.K.; Johne, R.; Wedemeyer, H.; Bock, C.T.; Kremsner, P.G.; Velavan, T.P. Hepatitis E Virus Genome Detection in Commercial Pork Livers and Pork Meat Products in Germany. J. Viral Hepat. 2020, 28, 196–204. [Google Scholar] [CrossRef]
- Locus, T.; Lambrecht, E.; Peeters, M.; Suin, V.; Verhaegen, B.; Van Hoorde, K.; Lamoral, S.; Vanwolleghem, T.; Van Gucht, S. Hepatitis E Virus in Pork Meat Products and Exposure Assessment in Belgium. Int. J. Food Microbiol. 2023, 397, 110198. [Google Scholar] [CrossRef]
- Milojević, L.; Velebit, B.; Teodorović, V.; Kirbiš, A.; Petrović, T.; Karabasil, N.; Dimitrijević, M. Screening and Molecular Characterization of Hepatitis E Virus in Slaughter Pigs in Serbia. Food Environ. Virol. 2019, 11, 410–419. [Google Scholar] [CrossRef]
- Dzierzon, J.; Oswaldi, V.; Merle, R.; Langkabel, N.; Meemken, D. Hepatitis E Virus Cross-Contamination on the Surface of Porcine Livers after Storage in Euro Meat Containers in a German Pig Abattoir. J. Verbraucherschutz Leb. 2022, 17, 33–39. [Google Scholar] [CrossRef]
- Berto, A.; Martelli, F.; Grierson, S.; Banks, M. Hepatitis e Virus in Pork Food Chain, United Kingdom, 2009–2010. Emerg. Infect. Dis. 2012, 18, 1358–1360. [Google Scholar] [CrossRef]
- Berto, A.; Van der Poel, W.H.M.; Hakze-van der Honing, R.; Martelli, F.; La Ragione, R.M.; Inglese, N.; Collins, J.; Grierson, S.; Johne, R.; Reetz, J.; et al. Replication of Hepatitis E Virus in Three-Dimensional Cell Culture. J. Virol. Methods 2013, 187, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Feagins, A.R.; Opriessnig, T.; Guenette, D.K.; Halbur, P.G.; Meng, X.J. Inactivation of Infectious Hepatitis E Virus Present in Commercial Pig Livers Sold in Local Grocery Stores in the United States. Int. J. Food Microbiol. 2008, 123, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Tanaka, T.; Jirintai, S.; Nagashima, S.; Takahashi, M.; Nishizawa, T.; Mizuo, H.; Yazaki, Y.; Okamoto, H. A549 and PLC/PRF/5 Cells Can Support the Efficient Propagation of Swine and Wild Boar Hepatitis E Virus (HEV) Strains: Demonstration of HEV Infectivity of Porcine Liver Sold as Food. Arch. Virol. 2012, 157, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Chijiwa, K.; Sera, N.; Ishibashi, T.; Etoh, Y.; Shinohara, Y.; Kurata, Y.; Ishida, M.; Sakamoto, S.; Takeda, N.; et al. Hepatitis E Virus Transmission from Wild Boar Meat. Emerg. Infect. Dis. 2005, 11, 1958. [Google Scholar] [CrossRef]
- Matsubayashi, K.; Kang, J.H.; Sakata, H.; Takahashi, K.; Shindo, M.; Kato, M.; Sato, S.; Kato, T.; Nishimori, H.; Tsuji, K.; et al. A Case of Transfusion-Transmitted Hepatitis e Caused by Blood from a Donor Infected with Hepatitis e Virus via Zoonotic Food-Borne Route. Transfusion 2008, 48, 1368–1375. [Google Scholar] [CrossRef]
- Deest, G.; Zehner, L.; Nicand, E.; Gaudy-Graffin, C.; Goudeau, A.; Bacq, Y. Hépatite Virale E Autochtone En France et Consommation de Viande de Porc Séchée. Gastroenterol. Clin. Biol. 2007, 31, 1095–1097. [Google Scholar] [CrossRef]
- Renou, C.; Afonso, A.M.R.; Pavio, N. Foodborne Transmission of Hepatitis E Virus from Raw Pork Liver Sausage, France. Emerg. Infect. Dis. 2014, 20, 1945–1947. [Google Scholar] [CrossRef]
- Johne, R.; Trojnar, E.; Filter, M.; Hofmann, J. Thermal Stability of Hepatitis E Virus as Estimated by a Cell Culture Method. Appl. Environ. Microbiol. 2016, 82, 4225–4231. [Google Scholar] [CrossRef]
- Wolff, A.; Günther, T.; Albert, T.; Johne, R. Effect of Sodium Chloride, Sodium Nitrite and Sodium Nitrate on the Infectivity of Hepatitis E Virus. Food Environ. Virol. 2020, 12, 350–354. [Google Scholar] [CrossRef]
- Wolff, A.; Günther, T.; Albert, T.; Schilling-Loeffler, K.; Gadicherla, A.K.; Johne, R. Stability of Hepatitis E Virus at Different PH Values. Int. J. Food Microbiol. 2020, 325, 108625. [Google Scholar] [CrossRef]
- Barnaud, E.; Rogée, S.; Garry, P.; Rose, N.; Pavio, N. Thermal Inactivation of Infectious Hepatitis E Virus in Experimentally Contaminated Food. Appl. Environ. Microbiol. 2012, 78, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, T.; Sugiyama, R.; Shiota, T.; Li, T.C.; Yoshizaki, S.; Wakita, T.; Ishii, K. Evaluation of Heating Conditions for Inactivation of Hepatitis e Virus Genotypes 3 and 4. J. Food Prot. 2018, 81, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell Culture Systems for the Study of Hepatitis E Virus. Antivir. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell Culture Models for Hepatitis E Virus. Viruses 2019, 11, 608. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; D’Agostino, M.; Johne, R. Potential Approaches to Assess the Infectivity of Hepatitis E Virus in Pork Products: A Review. Food Environ. Virol. 2017, 9, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Martin-Latil, S.; Hennechart-Collette, C.; Delannoy, S.; Guillier, L.; Fach, P.; Perelle, S. Quantification of Hepatitis E Virus in Naturally-Contaminated Pig Liver Products. Front. Microbiol. 2016, 7, 1183. [Google Scholar] [CrossRef] [PubMed]
- Martin-Latil, S.; Hennechart-Collette, C.; Guillier, L.; Perelle, S. Method for HEV Detection in Raw Pig Liver Products and Its Implementation for Naturally Contaminated Food. Int. J. Food Microbiol. 2014, 176, 1–8. [Google Scholar] [CrossRef]
- Di Bartolo, I.; Angeloni, G.; Ponterio, E.; Ostanello, F.; Ruggeri, F.M. Detection of Hepatitis E Virus in Pork Liver Sausages. Int. J. Food Microbiol. 2015, 193, 29–33. [Google Scholar] [CrossRef]
- Cook, N.; D’Agostino, M.; Clarke, E.; Johne, R. FSA Project FS301014: A Critical Review of Approaches to Assess the Infectivity of Hepatitis E Virus. A Report to the United Kingdom Food Standards Agency; Fera Science Ltd. (FERA): London, UK, 2016; pp. 1–72. [Google Scholar]
- Fraisse, A.; Niveau, F.; Hennechart-Collette, C.; Coudray-Meunier, C.; Martin-Latil, S.; Perelle, S. Discrimination of Infectious and Heat-Treated Norovirus by Combining Platinum Compounds and Real-Time RT-PCR. Int. J. Food Microbiol. 2018, 269, 64–74. [Google Scholar] [CrossRef]
- Ceuppens, S.; Li, D.; Uyttendaele, M.; Renault, P.; Ross, P.; Van Ranst, M.; Cocolin, L.; Donaghy, J. Molecular Methods in Food Safety Microbiology: Interpretation and Implications of Nucleic Acid Detection. Compr. Rev. Food Sci. Food Saf. 2014, 13, 551–577. [Google Scholar] [CrossRef]
- Cuevas-Ferrando, E.; Girón-Guzmán, I.; Falcó, I.; Pérez-Cataluña, A.; Díaz-Reolid, A.; Aznar, R.; Randazzo, W.; Sánchez, G. Discrimination of Non-Infectious SARS-CoV-2 Particles from Fomites by Viability RT-QPCR. Environ. Res. 2022, 203, 111831. [Google Scholar] [CrossRef]
- Raymond, P.; Paul, S.; Guy, R.A. Impact of Capsid and Genomic Integrity Tests on Norovirus Extraction Recovery Rates. Foods 2023, 12, 826. [Google Scholar] [CrossRef]
- Cuevas-Ferrando, E.; Randazzo, W.; Pérez-Cataluña, A.; Falcó, I.; Navarro, D.; Martin-Latil, S.; Díaz-Reolid, A.; Girón-Guzmán, I.; Allende, A.; Sánchez, G. Platinum Chloride-Based Viability RT-QPCR for SARS-CoV-2 Detection in Complex Samples. Sci. Rep. 2021, 11, 18120. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, W.; Vasquez-García, A.; Aznar, R.; Sánchez, G. Viability RT-QPCR to Distinguish between HEV and HAV with Intact and Altered Capsids. Front. Microbiol. 2018, 9, 1973. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, W.; López-Gálvez, F.; Allende, A.; Aznar, R.; Sánchez, G. Evaluation of Viability PCR Performance for Assessing Norovirus Infectivity in Fresh-Cut Vegetables and Irrigation Water. Int. J. Food Microbiol. 2016, 229, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, W.; Khezri, M.; Ollivier, J.; Le Guyader, F.S.; Rodríguez-Díaz, J.; Aznar, R.; Sánchez, G. Optimization of PMAxx Pretreatment to Distinguish between Human Norovirus with Intact and Altered Capsids in Shellfish and Sewage Samples. Int. J. Food Microbiol. 2018, 266, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, X.; Sánchez, G.; Randazzo, W. Viability RT-QPCR to Detect Potentially Infectious Enteric Viruses on Heat-Processed Berries. Food Control 2020, 107, 106818. [Google Scholar] [CrossRef]
- Leifels, M.; Dan, C.; Sozzi, E.; Shoults, D.C.; Wuertz, S.; Mongkolsuk, S.; Sirikanchana, K. Capsid Integrity Quantitative PCR to Determine Virus Infectivity in Environmental and Food Applications; a Systematic Review. medRxiv 2020. [Google Scholar] [CrossRef]
- Leifels, M.; Jurzik, L.; Wilhelm, M.; Hamza, I.A. Use of Ethidium Monoazide and Propidium Monoazide to Determine Viral Infectivity upon Inactivation by Heat, UV- Exposure and Chlorine. Int. J. Hyg. Environ. Health 2015, 218, 686–693. [Google Scholar] [CrossRef]
- Leifels, M.; Shoults, D.; Wiedemeyer, A.; Ashbolt, N.J.; Sozzi, E.; Hagemeier, A.; Jurzik, L. Capsid Integrity QPCR-an Azo-Dye Based and Culture-Independent Approach to Estimate Adenovirus Infectivity after Disinfection and in the Aquatic Environment. Water 2019, 11, 1196. [Google Scholar] [CrossRef]
- Baert, L.; Wobus, C.E.; Van Coillie, E.; Thackray, L.B.; Debevere, J.; Uyttendaele, M. Detection of Murine Norovirus 1 by Using Plaque Assay, Transfection Assay, and Real-Time Reverse Transcription-PCR before and after Heat Exposure. Appl. Environ. Microbiol. 2008, 74, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Schielke, A.; Filter, M.; Appel, B.; Johne, R. Thermal Stability of Hepatitis e Virus Assessed by a Molecular Biological Approach. Virol. J. 2011, 8, 487. [Google Scholar] [CrossRef] [PubMed]
- Mormann, S.; Dabisch, M.; Becker, B. Effects of Technological Processes on the Tenacity and Inactivation of Norovirus Genogroup II in Experimentally Contaminated Foods. Appl. Environ. Microbiol. 2010, 76, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.; Santos, R. Enzymatic and Viability RT-QPCR Assays for Evaluation of Enterovirus, Hepatitis A Virus and Norovirus Inactivation: Implications for Public Health Risk Assessment. J. Appl. Microbiol. 2018, 124, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.I.; Cross, L.J.; Stapleton, T.A.; Jenkins, C.L.; Lees, D.N.; Lowther, J.A. Assessment of the Applicability of Capsid-Integrity Assays for Detecting Infectious Norovirus Inactivated by Heat or UV Irradiation. Food Environ. Virol. 2019, 11, 229–237. [Google Scholar] [CrossRef]
- Oristo, S.; Lee, H.J.; Maunula, L. Performance of Pre-RT-QPCR Treatments to Discriminate Infectious Human Rotaviruses and Noroviruses from Heat-Inactivated Viruses: Applications of PMA/PMAxx, Benzonase and RNase. J. Appl. Microbiol. 2018, 124, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Rogée, S.; Talbot, N.; Caperna, T.; Bouquet, J.; Barnaud, E.; Pavio, N. New Models of Hepatitis E Virus Replication in Human and Porcine Hepatocyte Cell Lines. J. Gen. Virol. 2013, 94, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Marti, E.; Ferrary-Américo, M.; Barardi, C.R.M. Detection of Potential Infectious Enteric Viruses in Fresh Produce by (RT)-QPCR Preceded by Nuclease Treatment. Food Environ. Virol. 2017, 9, 444–452. [Google Scholar] [CrossRef]
- Wolf, S.; Rivera-Aban, M.; Greening, G.E. Long-Range Reverse Transcription as a Useful Tool to Assess the Genomic Integrity of Norovirus. Food Environ. Virol. 2009, 1, 129–136. [Google Scholar] [CrossRef]
- Li, D.; De Keuckelaere, A.; Uyttendaele, M. Application of Long-Range and Binding Reverse Transcription-Quantitative PCR to Indicate the Viral Integrities of Noroviruses. Appl. Environ. Microbiol. 2014, 80, 6473–6479. [Google Scholar] [CrossRef]
- Rockey, N.; Young, S.; Kohn, T.; Pecson, B.; Wobus, C.E.; Raskin, L.; Wigginton, K.R. UV Disinfection of Human Norovirus: Evaluating Infectivity Using a Genome-Wide PCR-Based Approach. Environ. Sci. Technol. 2020, 54, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Razafimahefa, R.M.; Ludwig-Begall, L.F.; Le Guyader, F.S.; Farnir, F.; Mauroy, A.; Thiry, E. Optimisation of a PMAxxTM-RT-QPCR Assay and the Preceding Extraction Method to Selectively Detect Infectious Murine Norovirus Particles in Mussels. Food Environ. Virol. 2021, 13, 93–106. [Google Scholar] [CrossRef]
- Rodríguez, R.A.; Bounty, S.; Linden, K.G. Long-Range Quantitative PCR for Determining Inactivation of Adenovirus 2 by Ultraviolet Light. J. Appl. Microbiol. 2013, 114, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenröder, J.; Nickel, P.; Hofmann, J. An ORF1-Rearranged Hepatitis e Virus Derived from a Chronically Infected Patient Efficiently Replicates in Cell Culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Schemmerer, M.; Apelt, S.; Trojnar, E.; Ulrich, R.G.; Wenzel, J.J.; Johne, R. Enhanced Replication of Hepatitis E Virus Strain 47832C in an A549-Derived Subclonal Cell Line. Viruses 2016, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Capelli, N.; Dubois, M.; Pucelle, M.; Da Silva, I.; Lhomme, S.; Abravanel, F.; Chapuy-Regaud, S.; Izopet, J. Optimized Hepatitis e Virus (HEV) Culture and Its Application to Measurements of HEV Infectivity. Viruses 2020, 12, 139. [Google Scholar] [CrossRef]
- Chew, N.; Situ, J.; Wu, S.; Yao, W.; Sridhar, S. Independent Evaluation of Cell Culture Systems for Hepatitis E Virus. Viruses 2022, 14, 1254. [Google Scholar] [CrossRef]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Pas, S.D.; de Man, R.A.; Mulders, C.; Balk, A.H.M.M.; van Hal, P.T.W.; Weimar, W.; Koopmans, M.P.G.; Osterhaus, A.D.M.E.; van der Eijk, A.A. Hepatitis E Virus Infection among Solid Organ Transplant Recipients, the Netherlands. Emerg. Infect. Dis. 2012, 18, 869–872. [Google Scholar] [CrossRef]
- Fogeda, M.; Avellón, A.; Cilla, C.G.; Echevarría, J.M. Imported and Autochthonous Hepatitis E Virus Strains in Spain. J. Med. Virol. 2009, 81, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust Hepatitis e Virus Infection and Transcriptional Response in Human Hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- van de Garde, M.; Pass, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) Genotype 3 Infection of Human Liver Chimeric Mice as a Model for Chronic HEV Infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Chisari, F.V. Production of Infectious Hepatitis C Virus by Well-Differentiated, Growth-Arrested Human Hepatoma-Derived Cells. J. Virol. 2006, 80, 10253–10257. [Google Scholar] [CrossRef] [PubMed]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial Release of Hepatitis E Virus in Polarized Human Hepatocytes. J. Virol. 2019, 93, e01207-18. [Google Scholar] [CrossRef]
- Belouzard, S.; Danneels, A.; Fénéant, L.; Séron, K.; Rouillé, Y.; Dubuisson, J. Entry and Release of Hepatitis C Virus in Polarized Human Hepatocytes. J. Virol. 2017, 91, e00478-17. [Google Scholar] [CrossRef]
- Sari, G.; Zhu, J.; Ambardekar, C.; Yin, X.; Boonstra, A.; Feng, Z.; Vanwolleghem, T. The Viral ORF3 Protein Is Required for Hepatitis E Virus Apical Release and Efficient Growth in Polarized Hepatocytes and Humanized Mice. J. Virol. 2021, 95, e00585-21. [Google Scholar] [CrossRef]
- Liu, S.; Yang, W.; Li, Y.; Sun, C. Fetal Bovine Serum, an Important Factor Affecting the Reproducibility of Cell Experiments. Sci. Rep. 2023, 13, 1942. [Google Scholar] [CrossRef]
- Ianiro, G.; Monini, M.; Ammendolia, M.G.; De Sabato, L.; Ostanello, F.; Vaccari, G.; Di Bartolo, I. In Vitro Replication of Swine Hepatitis E Virus (HEV): Production of Cell-Adapted Strains. Animals 2023, 13, 276. [Google Scholar] [CrossRef]
- Iki, S.; Yokota, S.I.; Okabayashi, T.; Yokosawa, N.; Nagata, K.; Fujii, N. Serum-Dependent Expression of Promyelocytic Leukemia Protein Suppresses Propagation of Influenza Virus. Virology 2005, 343, 106–115. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S.Y. The Soluble Serum Protein Gas6 Bridges Virion Envelope Phosphatidylserine to the TAM Receptor Tyrosine Kinase Axl to Mediate Viral Entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.L.; Ju, H.P.; Liu, Y.; Gao, T.T.; Wang, W.B.; Aurelian, L.; Zhao, P.; Qi, Z.T. Fetal Bovine Serum Inhibits Hepatitis C Virus Attachment to Host Cells. J. Virol. Methods 2013, 193, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Schilling-Loeffler, K.; Viera-Segura, O.; Corman, V.M.; Schneider, J.; Gadicherla, A.K.; Schotte, U.; Johne, R. Cell Culture Isolation and Whole Genome Characterization of Hepatitis e Virus Strains from Wild Boars in Germany. Microorganisms 2021, 9, 2302. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, N.; Keuling, O.; Becher, P.; Baechlein, C. Isolation of 15 Hepatitis E Virus Strains Lacking ORF1 Rearrangements from Wild Boar and Pig Organ Samples and Efficient Replication in Cell Culture. Transbound. Emerg. Dis. 2022, 69, e2617–e2628. [Google Scholar] [CrossRef]
- Li, D.; Baert, L.; Xia, M.; Zhong, W.; Van Coillie, E.; Jiang, X.; Uyttendaele, M. Evaluation of Methods Measuring the Capsid Integrity and/or Functions of Noroviruses by Heat Inactivation. J. Virol. Methods 2012, 181, 1–5. [Google Scholar] [CrossRef]
- Cuevas-ferrando, E.; Randazzo, W.; Pérez-cataluña, A.; Navarro, D.; Martin-latin, S.; Díaz-reolid, A.; Girón-guzmán, I.; Allende, A.; Sánchez, G. Viability RT-PCR for SARS-CoV-2: A Step Forward to Solve the Infectivity Quandary of Preservation and Food Safety Technologies; Institute of Agrochemistry and Food Technology, IATA-CSIC: Valencia, Spain, 2021. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Composition |
|---|---|
| MMEM-0 | Maintenance * MEM with 0% FBS |
| MMEM-2 | MMEM-0 + 2% FBS |
| MMEM-5 | MMEM-0 + 5% FBS |
| MMEM-10 | MMEM-0 + 10% FBS |
| MMEM-0-D | MMEM-0 + 2% DMSO |
| MMEM-2-D | MMEM-2 + 2% DMSO |
| MMEM-5-D | MMEM-5 + 2% DMSO |
| MMEM-10-D | MMEM-10 + 2% DMSO |
| MDMEM-0 | Maintenance * DMEM with 0% FBS |
| MDMEM-2 | MDMEM-0 + 2% FBS |
| MDMEM-5 | MDMEM-0 + 5% FBS |
| MDMEM-10 | MDMEM-0 + 10% FBS |
| MDMEM-0-D | MDMEM-0 + 2% DMSO |
| MDMEM-2-D | MDMEM-2 + 2% DMSO |
| MDMEM-5-D | MDMEM-5 + 2% DMSO |
| MDMEM-10-D | MDMEM-10 + 2% DMSO |
| Pair | Target | Forward (FW) Reverse (RV) Probe (Pb) | Sequence 5′-3′ | Start (Position) | Stop (Position) | Length (Nucleotides) |
|---|---|---|---|---|---|---|
| 1 | Pre-methyltransferase (MeT) | FW | CTACTGCCATTGAGCAGGC | 68 | 86 | 126 |
| RV | GCTGCCGGGGTTGCATC | 193 | 178 | |||
| Pb | CTGTGGTGGTTCGGCCGTT | 121 | 140 | |||
| 2 | Pre-methyltransferase (MeT) | FW | CTACTGCCATHGAGCARGC | 68 | 86 | 126 |
| RV | GCTGCCGGGGTTGCATC | 193 | 178 | |||
| Pb | CTGTGGTGGTTCGGCCGTT | 121 | 140 | |||
| 3 | Pre-methyltransferase (MeT) | FW | CTTGGCGAATGCTGTGGTG | 111 | 129 | 83 |
| RV | GCTGCCGGGGTTGCATC | 193 | 178 | |||
| 4 | Pre-methyltransferase (MeT) | FW | ACTACTGCCATTGAGCAGGC | 67 | 86 | 80 |
| RV | GATAAAAACGGCCGAACCACCAC | 146 | 124 | |||
| 5 | Pre-methyltransferase (MeT) | FW | GTGGTCGATGCCATGGAGG | 19 | 37 | 128 |
| RV | GATAAAAACGGCCGAACCACCAC | 146 | 124 | |||
| 6 | Y-domain & papain-like cysteine protease (PCP) | FW | CGYCAGCTYCAGTTTTATGC | 1306 | 1325 | 173 |
| RV | CAGGTRCACTCCTGCCC | 1478 | 1462 | |||
| 7 | Y-domain & papain-like cysteine protease (PCP) | FW | CGYCAGCTYCAGTTTTAYGC | 1306 | 1325 | 173 |
| RV | CAGGTRCACTCCTGCCC | 1478 | 1462 | |||
| 8 | methyltransferase (MeT) [73] | FW | CTCCTGGCRTYACWACTGC | 56 | 74 | 172 |
| RV | GGRTGRTTCCAIARVACYTC | 227 | 208 |
| Primer Pair | Concentration Forward Primer | Concentration Reverse Primer | MasterMix | Efficiency | Slope | Y-Intercept |
|---|---|---|---|---|---|---|
| 1 | 600 nM | 900 nM | TaqMan environmental | 1.971 | −3.394 | 33.75 |
| 6 | 250 nM | 250 nM | SSO advanced SYBR Green inhibitor tolerant | 1.971 | −3.393 | 32.24 |
| Parameter | PtCl4 | RNase A | Long-Range RT-qPCR | Cell Culture |
|---|---|---|---|---|
| Infectivity assessment | Indirect (Capsid integrity) | Indirect (Capsid integrity) | Indirect (genome integrity) | Direct |
| Time | +++ | +++ | ++ | + |
| User friendly | +++ | +++ | + | + |
| Cost price | +++ | ++ | + | + |
| Sensitivity | +++ | +++ | ++ | ++ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locus, T.; Lambrecht, E.; Lamoral, S.; Willems, S.; Van Gucht, S.; Vanwolleghem, T.; Peeters, M. A Multifaceted Approach for Evaluating Hepatitis E Virus Infectivity In Vitro: Cell Culture and Innovative Molecular Methods for Integrity Assessment. Vet. Sci. 2023, 10, 676. https://doi.org/10.3390/vetsci10120676
Locus T, Lambrecht E, Lamoral S, Willems S, Van Gucht S, Vanwolleghem T, Peeters M. A Multifaceted Approach for Evaluating Hepatitis E Virus Infectivity In Vitro: Cell Culture and Innovative Molecular Methods for Integrity Assessment. Veterinary Sciences. 2023; 10(12):676. https://doi.org/10.3390/vetsci10120676
Chicago/Turabian StyleLocus, Tatjana, Ellen Lambrecht, Sophie Lamoral, Sjarlotte Willems, Steven Van Gucht, Thomas Vanwolleghem, and Michael Peeters. 2023. "A Multifaceted Approach for Evaluating Hepatitis E Virus Infectivity In Vitro: Cell Culture and Innovative Molecular Methods for Integrity Assessment" Veterinary Sciences 10, no. 12: 676. https://doi.org/10.3390/vetsci10120676
APA StyleLocus, T., Lambrecht, E., Lamoral, S., Willems, S., Van Gucht, S., Vanwolleghem, T., & Peeters, M. (2023). A Multifaceted Approach for Evaluating Hepatitis E Virus Infectivity In Vitro: Cell Culture and Innovative Molecular Methods for Integrity Assessment. Veterinary Sciences, 10(12), 676. https://doi.org/10.3390/vetsci10120676