A Uniform In Vitro Efficacy Dataset to Guide Antimicrobial Peptide Design


Abstract
:1. Introduction
- Individual studies report MIC values obtained using varying protocols, which produce different results.
- Different groups use different type cultures of the same organism for MIC estimation.
- Negative data (MIC results for ineffective peptides) is seldom published.
2. Data Description
2.1. Minimum Inhibitory Concentration (MIC) Data
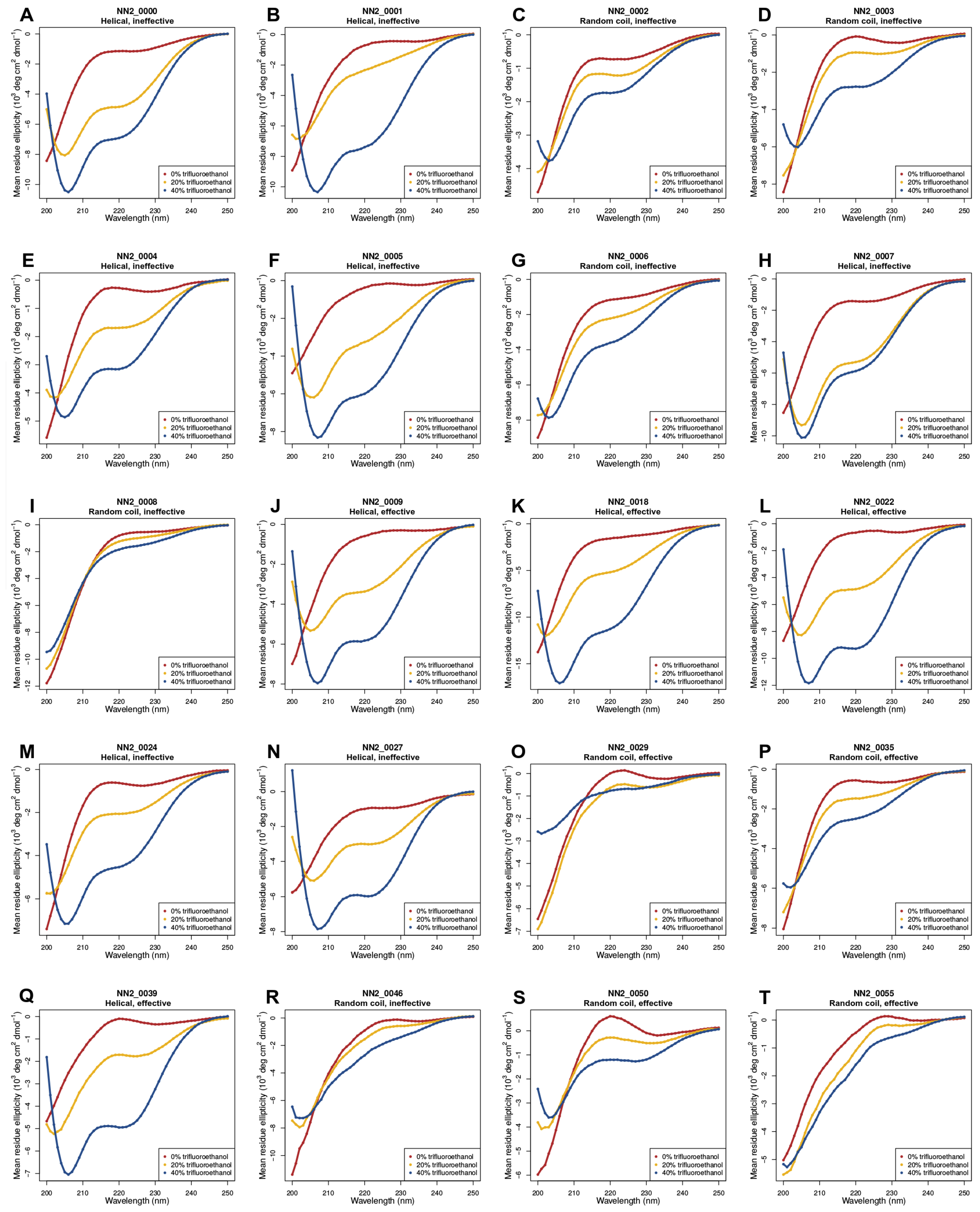
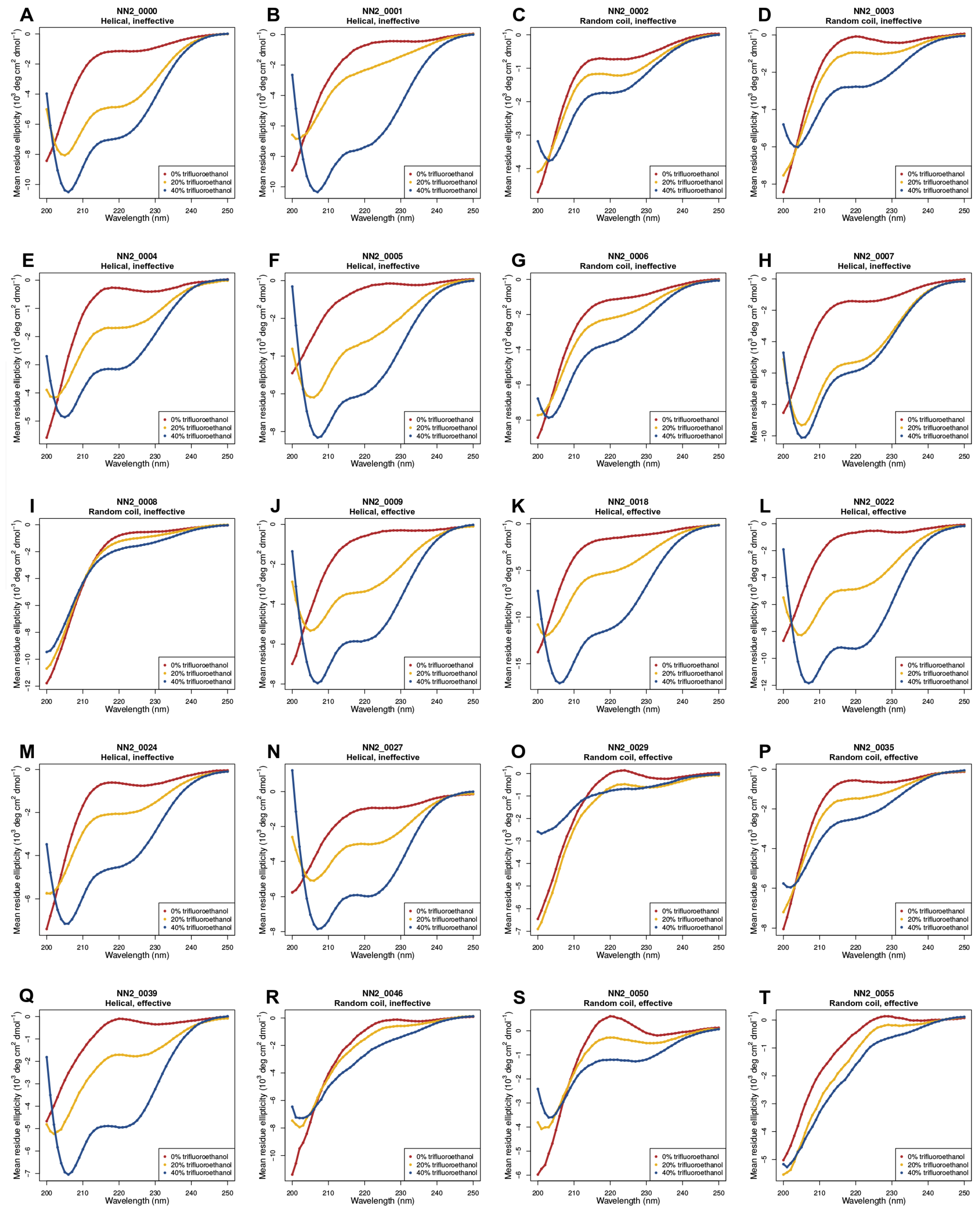
2.2. Circular Dichroism (CD) Data
3. Preliminary Analyses
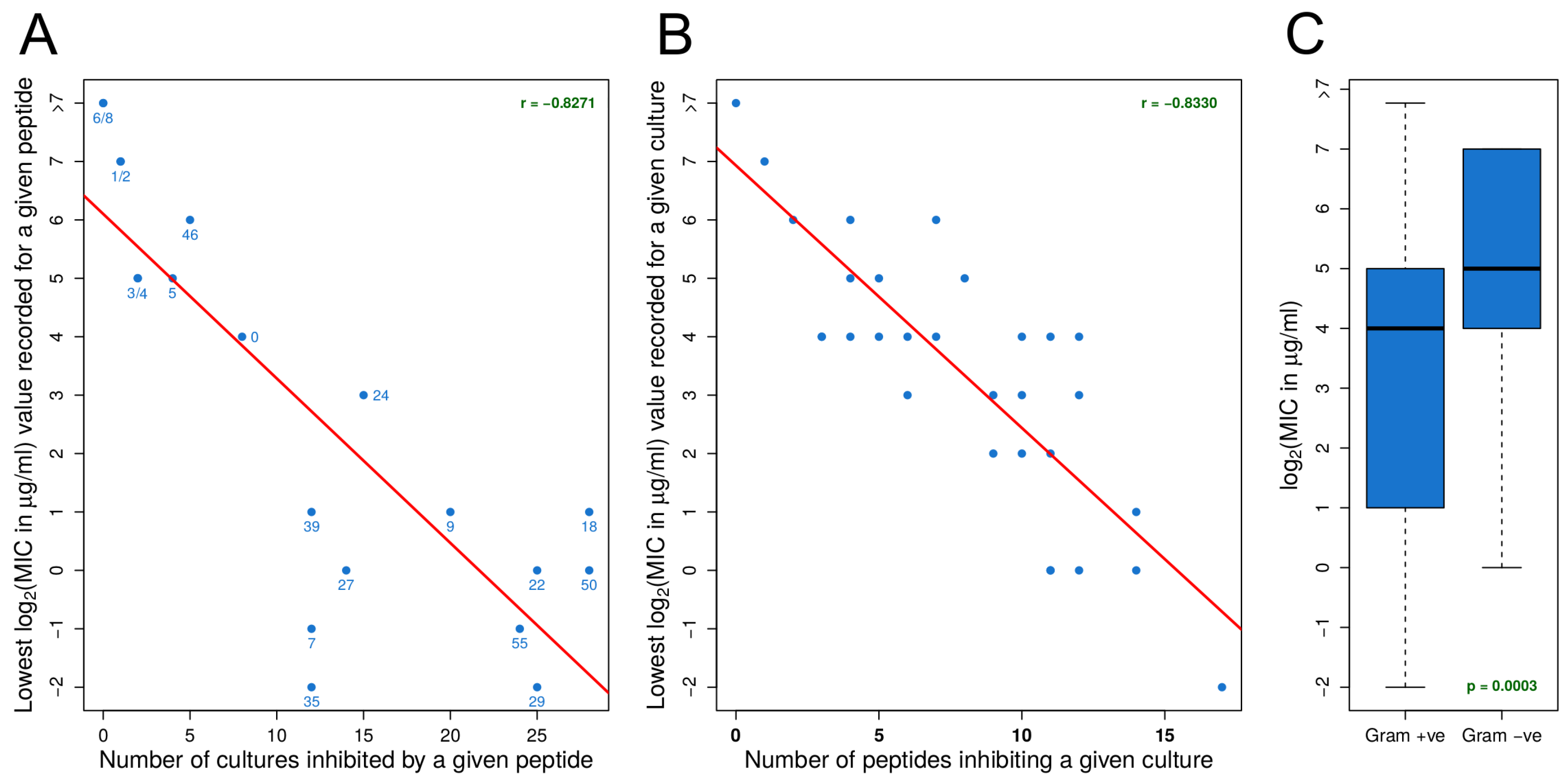
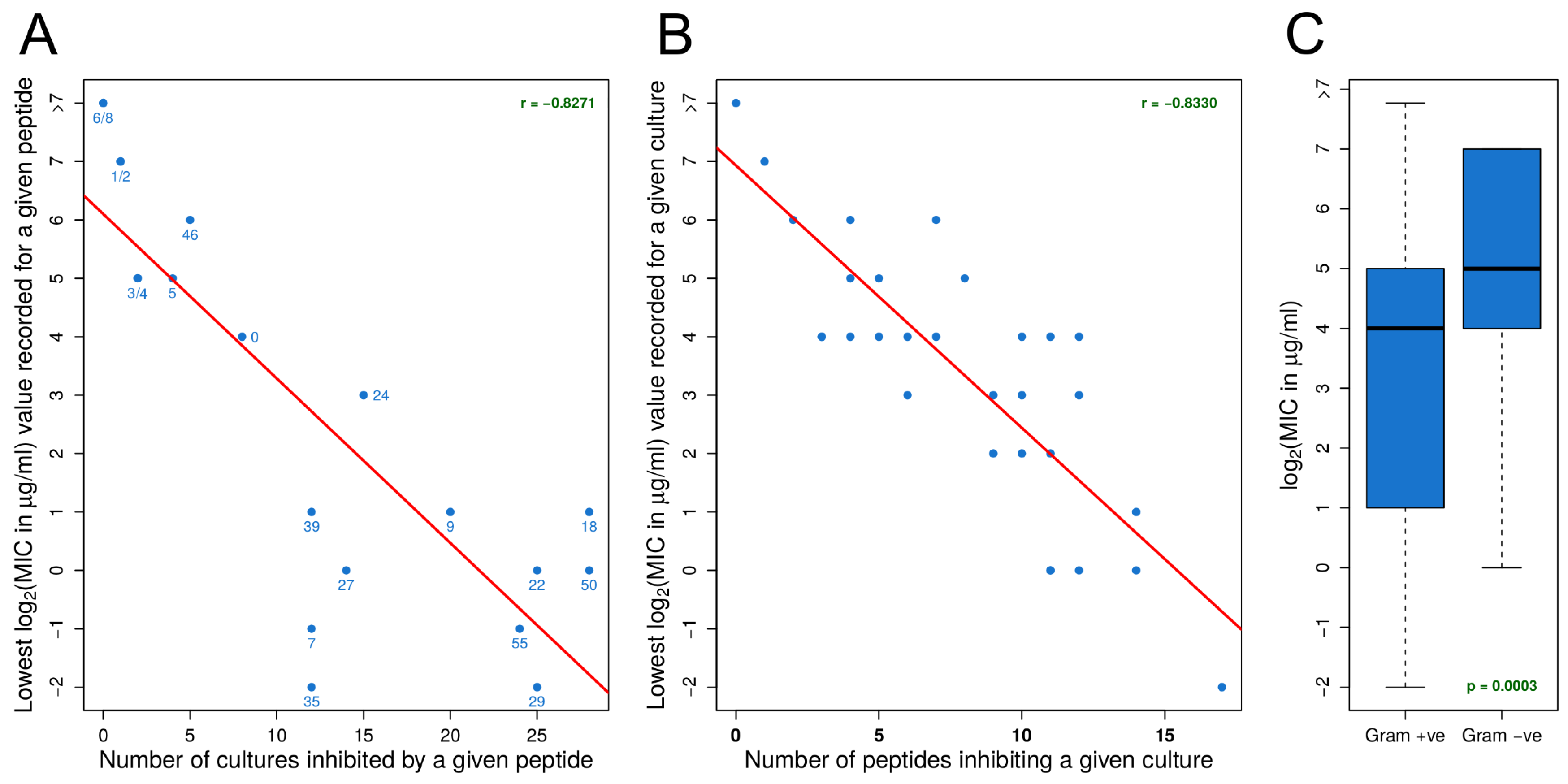
3.1. Identifying Effective Peptides Based on MIC Data
- X: a matrix of MIC values,
- M: rows containing MIC values for a given organism,
- N: columns containing MIC values for a given peptide,
- ,
- Multiple minimum MIC values can occur along a given row.
3.2. MIC Experiments Suggest a Common Mechanism of Action for Both Gram-Positive and Gram-Negative Organisms
- For an organism, susceptibility to one effective peptide indicates greater susceptibility to all effective peptides.
- For an effective peptide, efficacy for one organism indicates greater efficacy for all organisms.
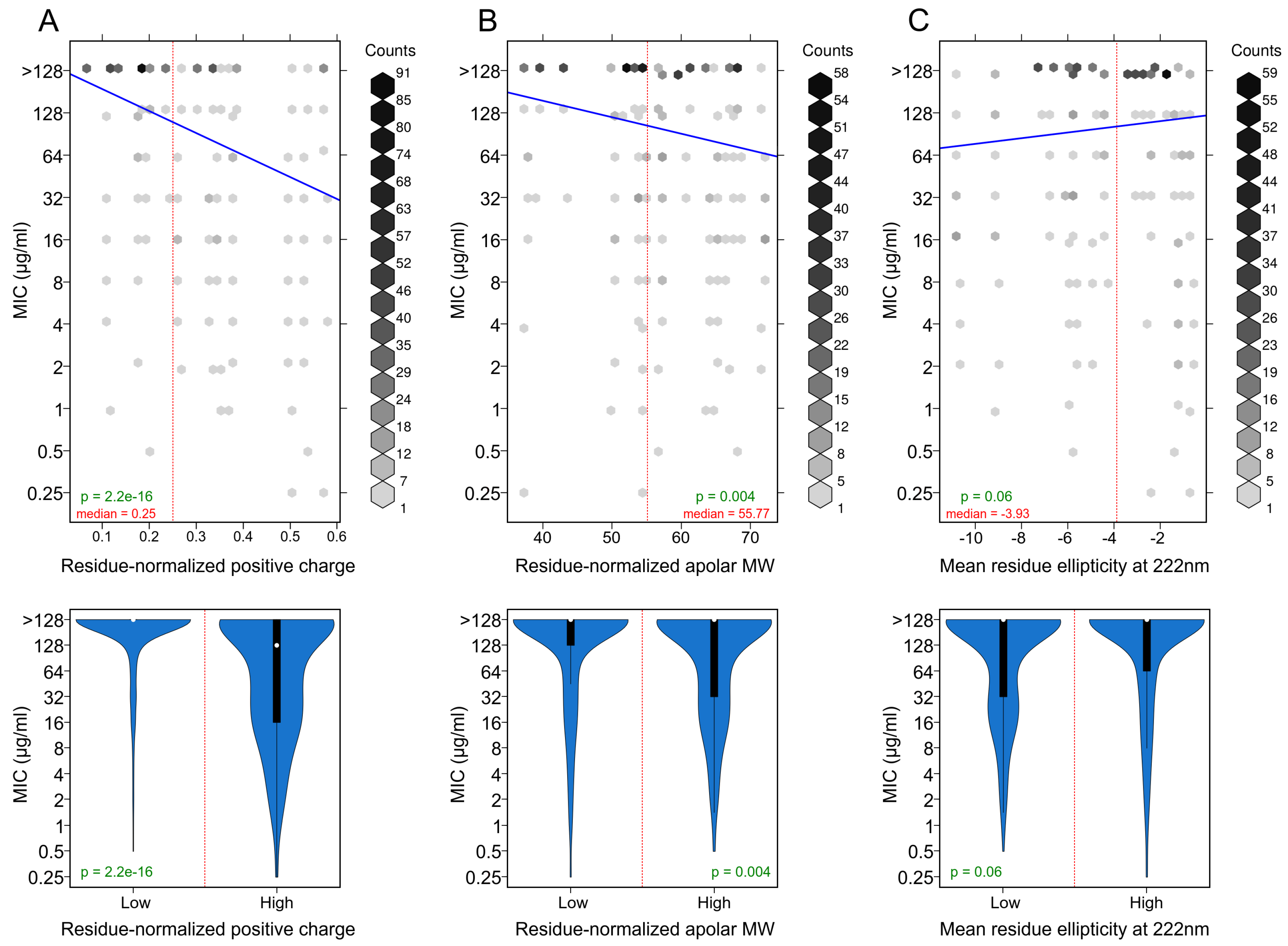
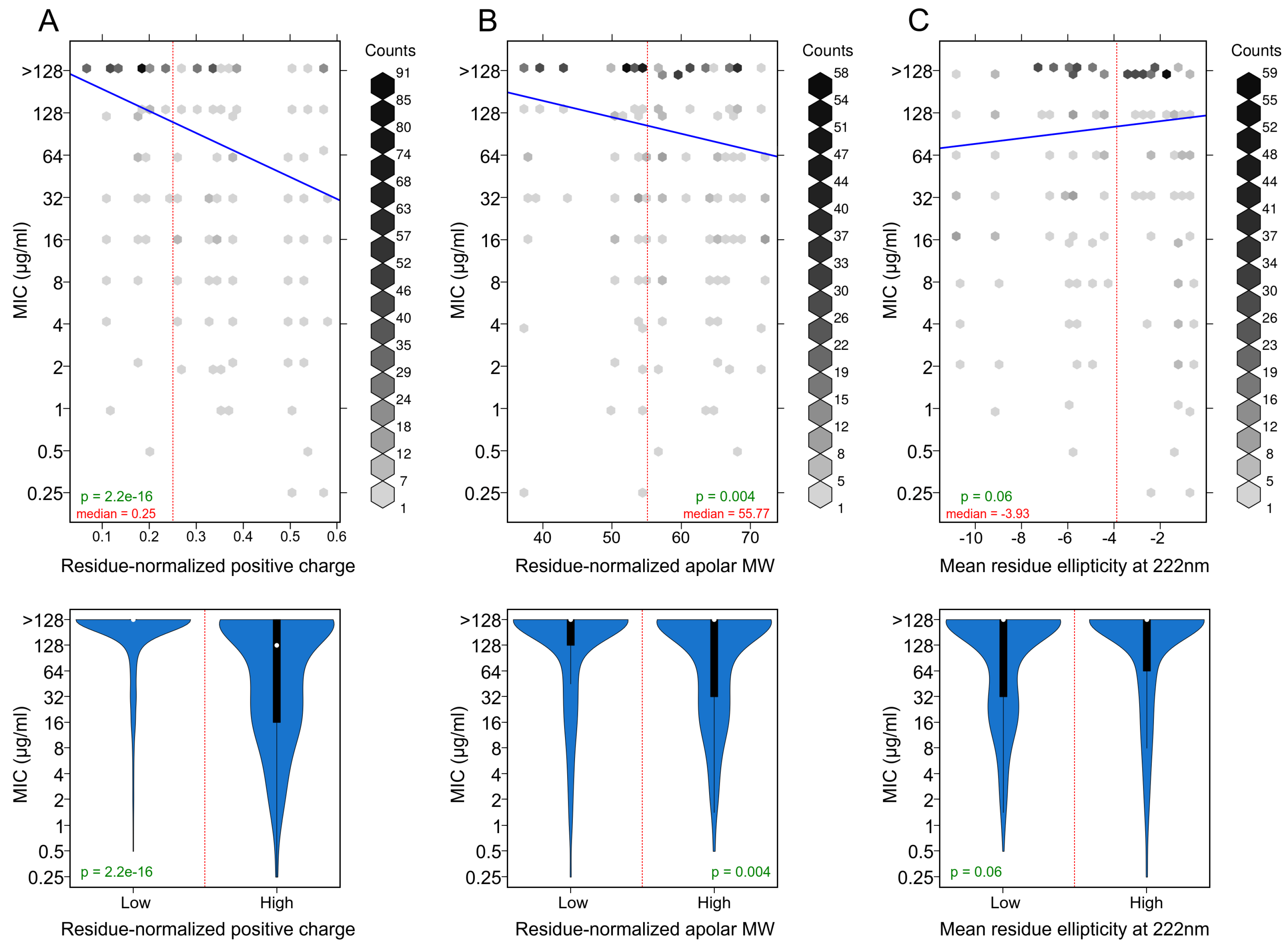
3.3. Positively Charged Residues Are Associated with Increased Peptide Activity
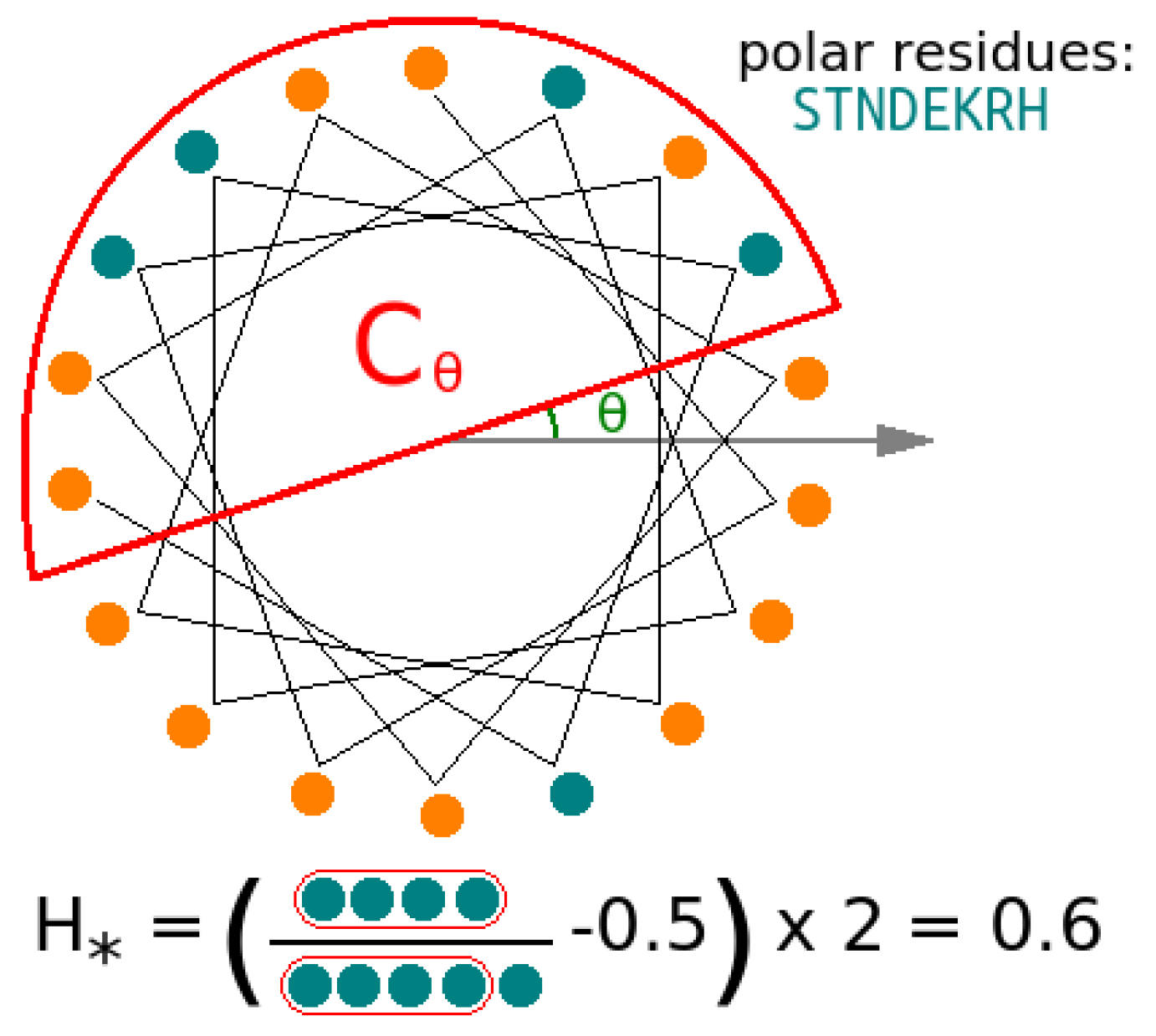
3.4. Apolar Residues Are Associated with Increased Peptide Activity
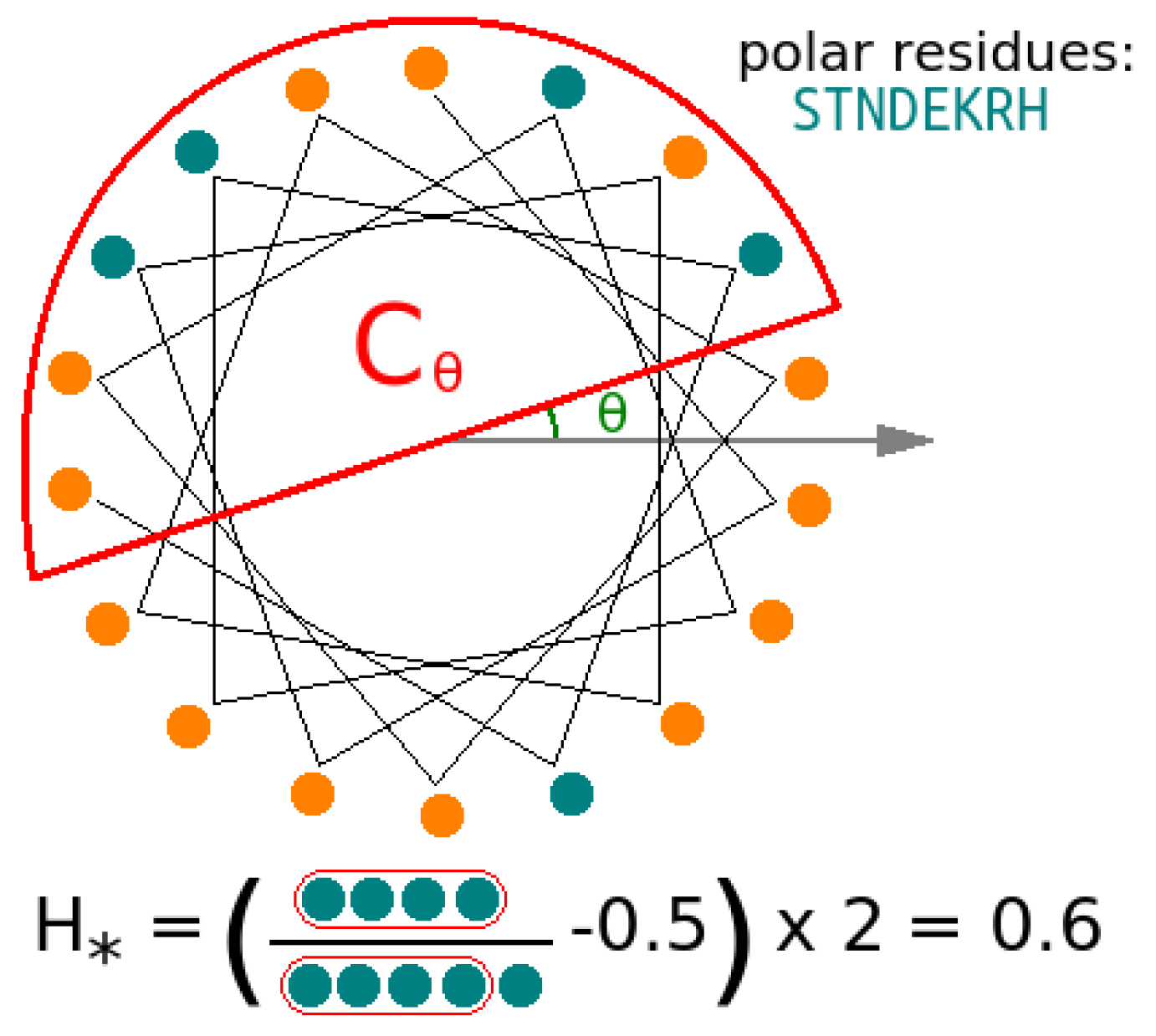
3.5. Helicity Is Not Essential for Peptide Activity
4. Discussion
- Our previous work [10] reported prominent blebbing observed on the S. haemolyticus cell membrane, and large-scale membrane damage observed on E. coli, upon treatment with peptides NN2_0018 and NN2_0050. These disruptions cannot be explained through the formation of nanometer-scale pores alone. Previously, the carpet model had successfully explained similar blebbing on the P. aeruginosa cell membrane [9].
5. Materials and Methods
5.1. Computational Design and Selection of Antimicrobial Peptides
5.2. Peptide Synthesis
5.3. Antimicrobial Susceptibility Assays
5.4. Circular Dichroism Experiments
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Marshall, J.C.; Namendys-Silva, S.A.; François, B.; Martin-Loeches, I.; Lipman, J.; Reinhart, K.; Antonelli, M.; Pickkers, P.; Njimi, H.; et al. Assessment of the worldwide burden of critical illness: The intensive care over nations (ICON) audit. Lancet Respir. Med. 2014, 2, 380–386. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, G. APD: The antimicrobial peptide database. Nucl. Acids Res. 2004, 32, D590–D592. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Hancock, R.E. Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit. Rev. Immunol. 2000, 20, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D.; Dubos, R.J. Bactericidal fractions from an aerobic sporulating bacillus. J. Biol. Chem. 1940, 136, 803–804. [Google Scholar]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.; Mueller, P. A molecular model of membrane excitability. J. Supramol. Struct. 1974, 2, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, D.; Nagarajan, T.; Roy, N.; Kulkarni, O.; Ravichandran, S.; Mishra, M.; Chakravortty, D.; Chandra, N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2018, 293, 3492–3509. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Kuwahara, J.; Fujii, N.; Sugiura, Y. Binding of tachyplesin I to DNA revealed by footprinting analysis: Significant contribution of secondary structure to DNA binding and implication for biological action. Biochemistry 1992, 31, 2998–3004. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 2002, 46, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Krämer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, E1409–E1418. [Google Scholar] [CrossRef] [PubMed]
- Gusman, H.; Travis, J.; Helmerhorst, E.J.; Potempa, J.; Troxler, R.F.; Oppenheim, F.G. Salivary histatin 5 is an inhibitor of both host and bacterial enzymes implicated in periodontal disease. Infect. Immun. 2001, 69, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Madera, L.; Ma, S.; Waldbrook, M.; Elliott, M.R.; Easton, D.M.; Mayer, M.L.; Mullaly, S.C.; Kindrachuk, J.; Jenssen, H.; et al. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J. Immunol. 2010, 184, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Befus, A.D.; Mowat, C.; Gilchrist, M.; Hu, J.; Solomon, S.; Bateman, A. Neutrophil defensins induce histamine secretion from mast cells: Mechanisms of action. J. Immunol. 1999, 163, 947–953. [Google Scholar] [PubMed]
- Koczulla, R.; von Degenfeld, G.; Kupatt, C.; Krötz, F.; Zahler, S.; Gloe, T.; Issbrücker, K.; Unterberger, P.; Zaiou, M.; Lebherz, C.; et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J. Clin. Investig. 2003, 111, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; MacDonald, D.L.; Holroyd, K.J.; Thornsberry, C.; Wexler, H.; Zasloff, M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999, 43, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Design of model amphipathic peptides having potent antimicrobial activities. Biochemistry 1992, 31, 12688–12694. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Steckbeck, J.D.; Craigo, J.K.; Doi, Y.; Mietzner, T.A.; Montelaro, R.C. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob. Agents Chemother. 2013, 57, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Phadke, S.M.; Lazarevic, V.; Cascio, M.; Islam, K.; Montelaro, R.C.; Mietzner, T.A. De novo generation of cationic antimicrobial peptides: Influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 2005, 49, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Wiradharma, N.; Khoe, U.; Hauser, C.A.; Seow, S.V.; Zhang, S.; Yang, Y.Y. Synthetic cationic amphiphilic α-helical peptides as antimicrobial agents. Biomaterials 2011, 32, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Cheung, W.A.; Hancock, R.E.; Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and cheminformatics. Chem. Biol. Drug Des. 2011, 77, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Maccari, G.; Di Luca, M.; Nifosí, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial peptides design by evolutionary multiobjective optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef] [PubMed]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucl. Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Gesell, J.; Zasloff, M.; Opella, S.J. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 1997, 9, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Kolodziejek, J.; Nowotny, N. Antimicrobial peptides from ranid frogs: Taxonomic and phylogenetic markers and a potential source of new therapeutic agents. Biochim. Biophys. Acta 2004, 1696, 1–14. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence |
|---|---|
| NN2_0000 | EVAKKLLASALKLALAI |
| NN2_0001 | EDWNHLGAAVHTLKHVYK |
| NN2_0002 | AIVEQLRKRC |
| NN2_0003 | KLSASLKHVAHRARHLS |
| NN2_0004 | ESRAGKLAAKAAFKAAKR |
| NN2_0005 | EWAAARQVIIHATRKY |
| NN2_0006 | EILSKALSALSPLAN |
| NN2_0007 | EKAILSALKLLRLAL |
| NN2_0008 | ETAKGVAKHLPPAIA |
| NN2_0009 | KVYARLHAVIKRLHRRLH |
| NN2_0018 | YLARAIRRTLARLLL |
| NN2_0022 | EWRVARRAVQRLRHLARRYH |
| NN2_0024 | ALKKMLRLAKRLS |
| NN2_0027 | VLSAFHKVIKIIHHISHF |
| NN2_0029 | RKFRKILHRARKWI |
| NN2_0035 | RRWGRWHRMRRRGR |
| NN2_0039 | FWKGLVKAAFKIVHAGS |
| NN2_0046 | GWKAIHKAAKGIHTYVN |
| NN2_0050 | SWKKFFKKARSLPKLF |
| NN2_0055 | YKRWKKWRSKAKKIL |
| Organism | Culture ID | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ | NN2_ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0000 | 0001 | 0002 | 0003 | 0004 | 0005 | 0006 | 0007 | 0008 | 0009 | 0018 | 0022 | 0024 | 0027 | 0029 | 0035 | 0039 | 0046 | 0050 | 0055 | ||
| E. coli | K12 MG1655 | 16 | 32 | 16 | 64 | 32 | 8 | 64 | 4 | 4 | |||||||||||
| A. baumanii | MTCC 9829 | 128 | 32 | 16 | 16 | 128 | 16 | 8 | 16 | 4 | 16 | ||||||||||
| S. boydii | MTCC 11947 | 128 | 128 | 8 | 32 | 8 | 64 | 16 | 1 | 64 | 2 | 8 | |||||||||
| S. flexnerii | MTCC 1457 | 128 | 128 | 4 | 8 | 1 | 32 | 8 | 4 | 64 | 4 | 8 | |||||||||
| S. typhimurium | ATCC 14028 | 32 | 32 | 16 | 16 | 8 | 32 | ||||||||||||||
| S. enterica | MTCC 9844 | 128 | 32 | 16 | 16 | 32 | 32 | 16 | 8 | 16 | |||||||||||
| K. pneumoniae | MTCC 7407 | 32 | 128 | 64 | 32 | 128 | |||||||||||||||
| K. oxytoca | MTCC 2275 | 128 | 8 | 16 | 8 | 128 | 32 | 64 | 64 | 16 | 32 | ||||||||||
| P. aeruginosa | MTCC 3542 | 128 | 16 | 32 | 128 | ||||||||||||||||
| P. vulgaris | MTCC 1771 | 128 | 128 | 64 | 16 | 64 | |||||||||||||||
| P. mirabilis | MTCC 3158 | ||||||||||||||||||||
| C. koserii | MTCC 1657 | 128 | 128 | 32 | 16 | 16 | 64 | 16 | 64 | 16 | 64 | 8 | 16 | ||||||||
| C. freundii | MTCC 1658 | 32 | 16 | 64 | 32 | 32 | 128 | ||||||||||||||
| N. mucosa * | MTCC 1772 | 128 | 128 | 32 | 16 | 32 | 128 | 32 | 32 | 64 | 128 | 16 | 64 | ||||||||
| V. cholerae | MTCC 3904 | 128 | 64 | 128 | 128 | 128 | 64 | 128 | |||||||||||||
| E. gergoviae | MTCC 3826 | 128 | 128 | 128 | 64 | ||||||||||||||||
| H. influenzae | MTCC 621 | 64 | 128 | 128 | 16 | 4 | 8 | 8 | 64 | 8 | 8 | 128 | 64 | 2 | 32 | ||||||
| A. fecalis | MTCC 1937 | 32 | 128 | 128 | 64 | ||||||||||||||||
| B. bronchiseptica | MTCC 6837 | 16 | 64 | 4 | 4 | 2 | 8 | 4 | 1 | 8 | 128 | 1 | 8 | ||||||||
| E. aerogenes | MTCC 111 | 32 | 16 | 128 | |||||||||||||||||
| S. maltophilia | MTCC 1890 | 128 | 16 | 64 | 128 | 128 | 16 | 128 | |||||||||||||
| M. luteus * | MTCC 425 | 32 | 128 | 32 | 32 | 32 | 0.5 | 2 | 2 | 2 | 8 | 1 | 0.25 | 0.25 | 2 | 64 | 2 | 0.5 | |||
| S. aureus | MTCC 3160 | 16 | 128 | 128 | |||||||||||||||||
| S. hemolyticus | MTCC 3383 | 128 | 4 | 16 | 16 | 128 | 32 | 8 | 4 | 16 | 8 | 4 | |||||||||
| E. faecalis | MTCC 439 | 64 | 128 | ||||||||||||||||||
| C. glutamicum | MTCC 2679 | 32 | 64 | 32 | 2 | 4 | 1 | 16 | 4 | 2 | 4 | 2 | 64 | 2 | 2 | ||||||
| C. pseudoTB * | MTCC 3158 | 128 | |||||||||||||||||||
| B. alcalophilis | MTCC 860 | 64 | 32 | 16 | 16 | 32 | 64 | 32 | 16 | 32 | 64 | ||||||||||
| M. smegmatis * | MC2155 | 128 | 32 | 64 | 16 | 64 | 32 | 16 | 16 | 128 | 64 | 32 | |||||||||
| C. albicans * | MTCC 425 | 32 | 128 | 64 | 128 | 64 | 64 | 64 | 64 | ||||||||||||
| net charge | 3 | 2 | 3 | 4 | 6 | 3 | 1 | 3 | 2 | 6 | 4 | 7 | 5 | 2 | 7 | 8 | 3 | 3 | 6 | 8 | |
| peptide score | 3 | 10 | 5 | 6 | 3 | 1 | 14 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagarajan, D.; Nagarajan, T.; Nanajkar, N.; Chandra, N. A Uniform In Vitro Efficacy Dataset to Guide Antimicrobial Peptide Design. Data 2019, 4, 27. https://doi.org/10.3390/data4010027
Nagarajan D, Nagarajan T, Nanajkar N, Chandra N. A Uniform In Vitro Efficacy Dataset to Guide Antimicrobial Peptide Design. Data. 2019; 4(1):27. https://doi.org/10.3390/data4010027
Chicago/Turabian StyleNagarajan, Deepesh, Tushar Nagarajan, Neha Nanajkar, and Nagasuma Chandra. 2019. "A Uniform In Vitro Efficacy Dataset to Guide Antimicrobial Peptide Design" Data 4, no. 1: 27. https://doi.org/10.3390/data4010027
APA StyleNagarajan, D., Nagarajan, T., Nanajkar, N., & Chandra, N. (2019). A Uniform In Vitro Efficacy Dataset to Guide Antimicrobial Peptide Design. Data, 4(1), 27. https://doi.org/10.3390/data4010027