RetroTransformDB: A Dataset of Generic Transforms for Retrosynthetic Analysis


Abstract
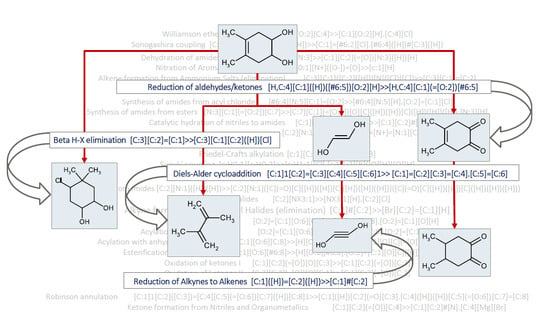
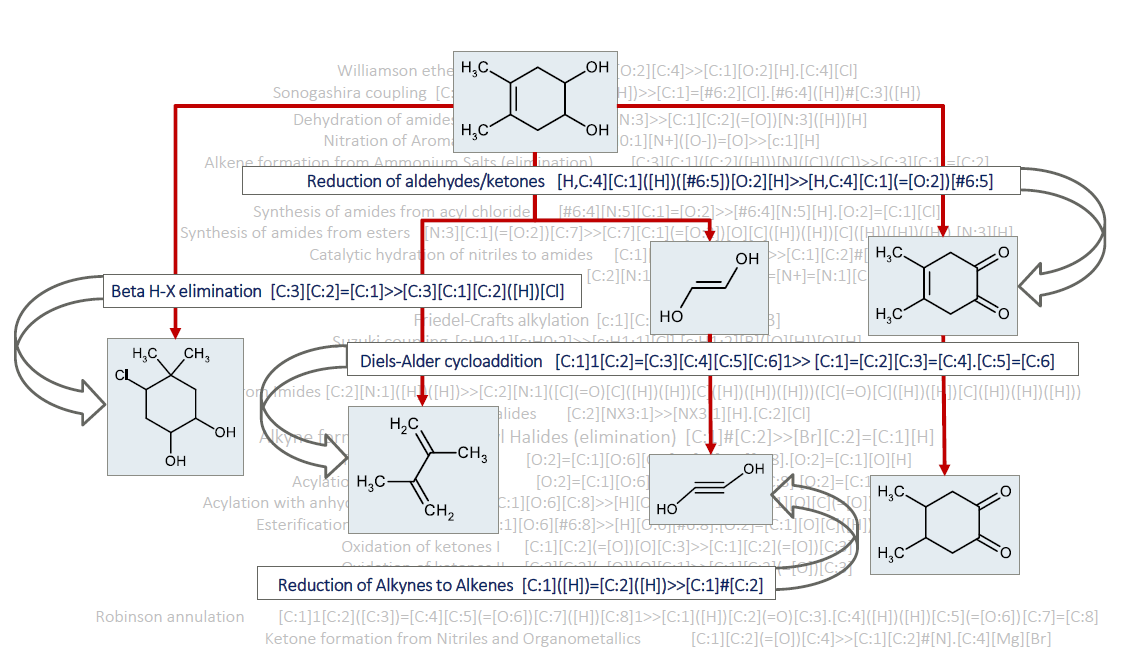{kind=link}
{kind=link}
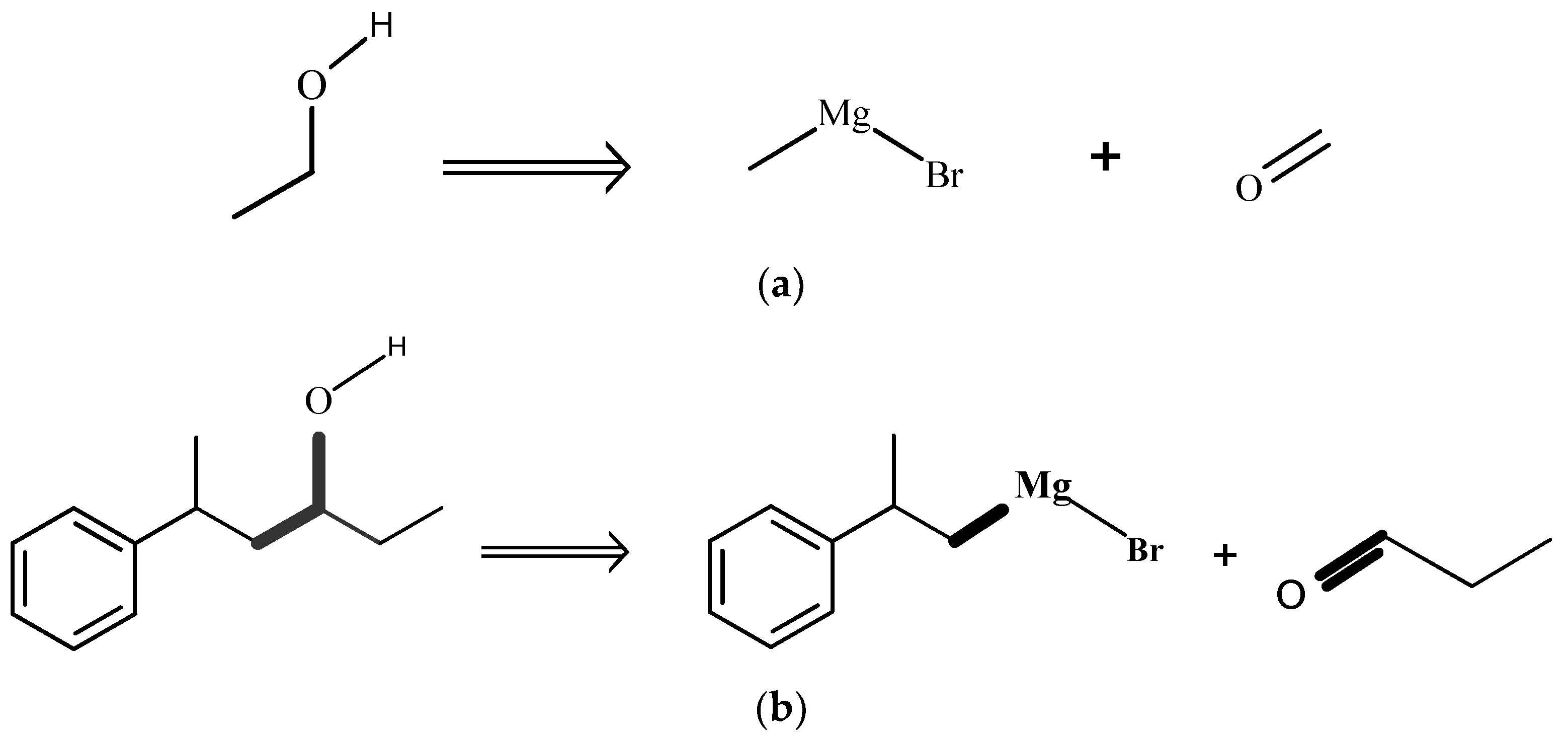{kind=link}
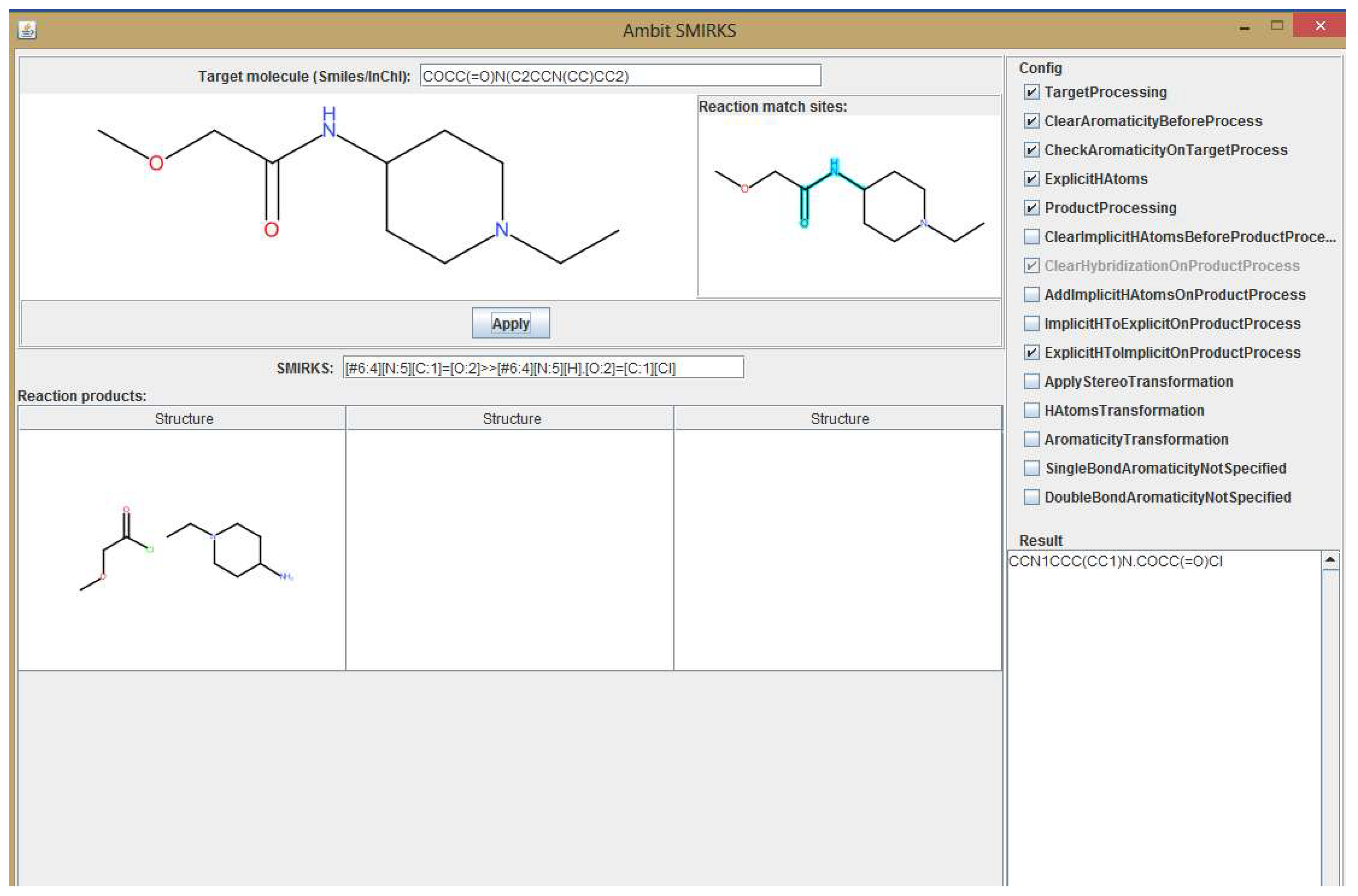{kind=link}
1. Summary
2. Data Description
3. Methods
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Baskin, I.I.; Madzhidov, T.I.; Antipin, I.S.; Varnek, A.A. Artificial intelligence in synthetic chemistry: Achievements and prospects. Russ. Chem. Rev. 2017, 86, 1127–1156. [Google Scholar] [CrossRef]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Nguyen, Q.L.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. A Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Zsoldos, Z.; Simon, A.; Reid, D.; Liu, Y.; Khew, S.Y.; Johnson, A.P.; Major, S.; Wade, R.A.; Ando, H.Y. Route Designer: A retrosynthetic analysis tool utilizing automated retrosynthetic rule generation. J. Chem. Inf. Model. 2009, 49, 593–602. [Google Scholar] [CrossRef] [PubMed]
- CMBI—LHASA. Available online: http://cheminf.cmbi.ru.nl/cheminf/olp/history.shtml (accessed on 2 February 2018).
- Wipke, W.T.; Braun, H.; Smith, G.; Choplin, F.; Sieber, W. SECS-Simulation and Evaluation of Chemical Synthesis: Strategy and Planning. In Computer-Assisted Organic Synthesis; ACS Publications: Washington, DC, USA, 1977; pp. 97–127. [Google Scholar]
- Krebsbach, D.; Gelernter, H.; Sieburth, S.M.N. Distributed heuristic synthesis search. J. Chem. Inf. Comput. Sci. 1998, 38, 595–604. [Google Scholar] [CrossRef]
- Tanaka, A.; Okamoto, H.; Bersohn, M. Construction of Functional Group Reactivity Database under Various Reaction Conditions Automatically Extracted from Reaction Database in a Synthesis Design System. J. Chem. Inf. Model. 2010, 50, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, L.L.; Yang, S.Y. RASA: A rapid retrosynthesis-based scoring method for the assessment of synthetic accessibility of drug-like molecules. J. Chem. Inf. Model. 2011, 51, 2768–2777. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Funatsu, K. A Novel Approach to Retrosynthetic Analysis Using Knowledge Bases Derived from Reaction Databases. J. Chem. Inf. Model. 1999, 39, 316–325. [Google Scholar]
- Chen, J.H.; Baldi, P. No Electron Left Behind: A Rule-Based Expert System To Predict Chemical Reactions and Reaction Mechanisms. J. Chem. Inf. Model. 2009, 49, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Elsevier, Reaxys Syntheis Planner. Available online: https://www.elsevier.com/solutions/reaxys/how-reaxys-works/synthesis-planner (accessed on 11 February 2018).
- Reactions—CASREACT. Available online: http://support.cas.org/content/reactions (accessed on 14 January 2018).
- InfoChem—SPRESI—Storage and Retrieval of Chemical Structure and Reaction Information. Available online: http://www.infochem.de/products/databases/spresi.shtml (accessed on 14 January 2018).
- Chen, L.; Nourse, J.G.; Christie, B.D.; Leland, B.A.; Grier, D.L. Over 20 Years of Reaction Access Systems from MDL: A Novel Reaction Substructure Search Algorithm. J. Chem. Inf. Comput. Sci. 2002, 42, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Daniel Lowe, Chemical Reactions from US Patents (1976–Sep 2016). Available online: https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873 (accessed on 12 April 2018).
- Hu, Y.; Bajorath, J. Chemical Transformations That Yield Compounds with Distinct Activity Profiles. ACS Med. Chem. Lett. 2011, 2, 523–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, Y.; Bajorath, J. Hierarchical Analysis of Bioactive Matched Molecular Pairs, Encoded Chemical Transformations, and Associated Substructures. Mol. Inform. 2016, 35, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Chen, D.Z.; Taylor, K.T. Automatic reaction mapping and reaction center detection. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 560–593. [Google Scholar] [CrossRef]
- Szymkuć, S.; Gajewska, E.P.; Klucznik, T.; Molga, K.; Dittwald, P.; Startek, M.; Bajczyk, M.; Grzybowski, B.A. Computer-Assisted Synthetic Planning: The End of the Beginning. Angew. Chem. Int. Ed. 2016, 55, 5904–5937. [Google Scholar] [CrossRef] [PubMed]
- Klucznik, T.; Mikulak-Klucznik, B.; McCormack, M.P.; Lima, H.; Szymkuć, S.; Bhowmick, M.; Molga, K.; Zhou, Y.; Rickershauser, L.; Gajewska, E.P.; et al. Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory. Chem 2018, 4, 522–532. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Eberle, M.; Meier, P.; Nieto-Oberhuber, C.; Altmann, K.-H.; Schneider, G.; Jacoby, E.; Renner, S. A Collection of Robust Organic Synthesis Reactions for In Silico Molecule Design. J. Chem. Inf. Model. 2011, 51, 3093–3098. [Google Scholar] [CrossRef] [PubMed]
- Hartenfeller, M.; Zettl, H.; Walter, M.; Rupp, M.; Reisen, F.; Proschak, E.; Weggen, S.; Stark, H.; Schneider, G. Dogs: Reaction-driven de novo design of bioactive compounds. PLoS Comput. Biol. 2012, 8, e1002380. [Google Scholar] [CrossRef] [PubMed]
- Masek, B.B.; Baker, D.S.; Dorfman, R.J.; Dubrucq, K.; Francis, V.C.; Nagy, S.; Richey, B.L.; Soltanshahi, F. Multistep Reaction Based de Novo Drug Design: Generating Synthetically Feasible Design Ideas. J. Chem. Inf. Model. 2016, 56, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Schürer, S.C.; Tyagi, P.; Muskal, S.M. Prospective exploration of synthetically feasible, medicinally relevant chemical space. J. Chem. Inf. Model. 2005, 45, 239–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, R.A.Y.; Bunnett, J.F. Nomenclature for organic chemical transformations (Recommendations 1988). Pure Appl. Chem. 1989, 61, 725–768. [Google Scholar] [CrossRef]
- Corey, E.J. The Logic of Chemical Synthesis; John Wiley & Sons: Toronto, ON, Canada, 1989. [Google Scholar]
- Jeliazkova, N.; Kochev, N.; Jeliazkov, V. ambitcli-3.0.2. 14 April 2016. Available online: https://zenodo.org/record/173560#.WjlcRyvfHVq (accessed on 19 December 2017).
- Ideaconsult Ltd., AMBIT. Available online: http://ambit.sourceforge.net/ (accessed on 19 December 2017).
- Jeliazkova, N.; Jeliazkov, V. AMBIT RESTful web services: An implementation of the OpenTox application programming interface. J. Cheminform. 2011, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Daylight, SMIRKS: A Reaction Transform Language. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed on 20 December 2017).
- Daylight, Reaction Toolkit. Available online: http://www.daylight.com/products/reaction_kit.html (accessed on 20 September 2017).
- Daylight, SMARTS: A Language for Describing Molecular Patterns. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 19 September 2017).
- Angelo, J.D.; Smith, M.B. Hybrid Retrosynthesis; Elsevier: New York, NY, USA, 2015. [Google Scholar]
- Ideaconsult Ltd. Ambit-SMIRKS. Available online: http://ambit.sourceforge.net/smirks.html (accessed on 20 April 2018).



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avramova, S.; Kochev, N.; Angelov, P. RetroTransformDB: A Dataset of Generic Transforms for Retrosynthetic Analysis. Data 2018, 3, 14. https://doi.org/10.3390/data3020014
Avramova S, Kochev N, Angelov P. RetroTransformDB: A Dataset of Generic Transforms for Retrosynthetic Analysis. Data. 2018; 3(2):14. https://doi.org/10.3390/data3020014
Chicago/Turabian StyleAvramova, Svetlana, Nikolay Kochev, and Plamen Angelov. 2018. "RetroTransformDB: A Dataset of Generic Transforms for Retrosynthetic Analysis" Data 3, no. 2: 14. https://doi.org/10.3390/data3020014
APA StyleAvramova, S., Kochev, N., & Angelov, P. (2018). RetroTransformDB: A Dataset of Generic Transforms for Retrosynthetic Analysis. Data, 3(2), 14. https://doi.org/10.3390/data3020014