Simultaneous Determination of AFB1 and AFM1 in Milk Samples by Ultra High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Sampling
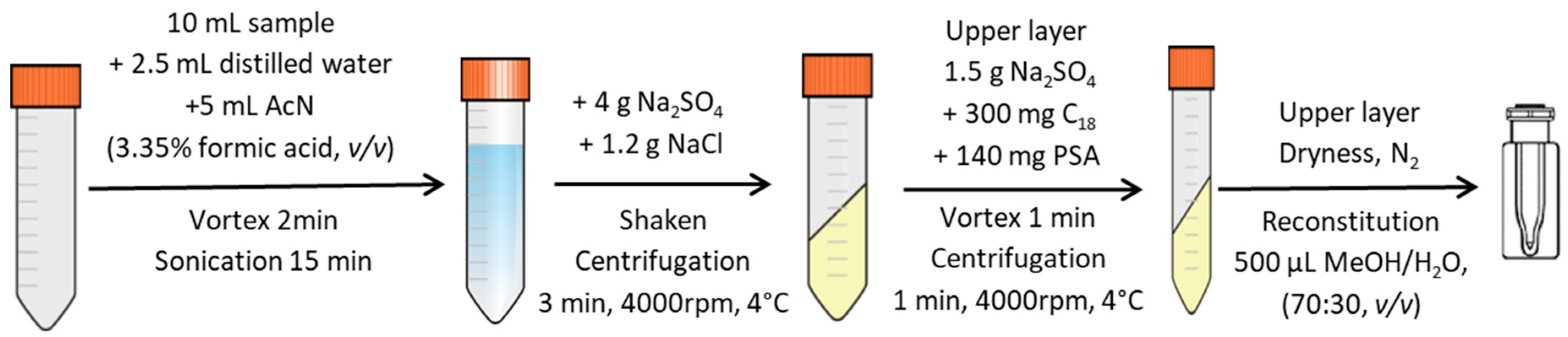
2.3. Sample Preparation
2.4. UHPLC-Q-Orbitrap HRMS Analysis
2.5. Validation of the Method
2.6. Quality Assurance/Quality Control (QA/QC) in the Analysis of Real Samples
3. Results and Discussion
3.1. Analytical Features of the Proposed Method
3.1.1. Linearity
3.1.2. Matrix Effect
3.1.3. Trueness and Precision
3.1.4. Specificity
3.1.5. Limits of Detection and Limits of Quantification
3.1.6. QA/QC
3.2. Application to Samples
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bhat, R.; Rai, R.V.; Karim, A.A. Mycotoxins in food and feed: Present status and future concerns. Compr. Rev. Food Sci. Food Saf. 2010, 9, 57–81. [Google Scholar] [CrossRef]
- Food and Agriculture Organization. Worldwide Regulations for Mycotoxins in Food and Feed in 2003, 2004. Available online: www.fao.org/docrep/007/y5499e/y5499e00.htm (accessed on 17 April 2018).
- International Agency for Research on Cancer. Agents Classified by the IARC Monographs; IARC: Lyon, France, 2012; Volume 1, p. 103. [Google Scholar]
- Commission Directive 2003/100/EC of 31 October 2003 Amending Annex I of Directive 2002/32/EC of the European Parliament and of the Council on Undesirable Substances in Animal Feed. 2003. Available online: https://eur-lex.europa.eu/eli/dir/2003/100/oj (accessed on 4 April 2018).
- International Agency for Research on Cancer, IARC. Evaluation of carcinogenic risks of chemical to humans. In Some Naturally-Occurring Substances: Food Items and Constituents; Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993; Volume 56, p. 245. [Google Scholar]
- Ketney, O.; Santini, A.; Oancea, S. Recent aflatoxin survey data in milk and milk products: A review. Int. J. Dairy Technol. 2017, 320–331. [Google Scholar] [CrossRef]
- Commission Regulation (EU) No 165/2010 of February 2010 Amending Regulation (EC) No 1881/2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs as Regards Aflatoxins. 2010. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32010R0165 (accessed on 4 April 2018).
- Food and Agriculture Organization Food Balance Sheet. 2013. Available online: http://www.fao.org/faostat/en/#data/FBS (accessed on 17 April 2018).
- Chen, C.Y.; Li, W.J.; Peng, K.Y. Determination of Aflatoxin M1 in Milk and Milk Powder Using High-Flow Solid-Phase Extraction and Liquid Chromatography—Tandem Mass Spectrometry. J. Agric. Food Chem. 2005, 53, 8474–8480. [Google Scholar] [CrossRef] [PubMed]
- Bognanno, M.; La Fauci, L.; Ritieni, A.; Tafuri, A.; De Lorenzo, A.; Micari, P.; Galvano, F. Survey of the occurrence of Aflatoxin M1 in ovine milk by HPLC and its confirmation by MS. Mol. Nutr. Food Res. 2006, 50, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Zinedine, A.; González-Osnaya, L.; Soriano, J.M.; Moltó, J.C.; Idrissi, L.; Mañes, J. Presence of aflatoxin M1 in pasteurized milk from Morocco. Int. J. Food Microbiol. 2007, 114, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Frenich, A.G.; Romero-González, R.; Gómez-Pérez, M.L.; Vidal, J.L.M. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, J.; Graziani, G.; Gaspari, A.; Chianese, D.; Ferrer, E.; Mañes, J.; Ritieni, A. Multi-Mycotoxin Analysis in Durum Wheat Pasta by Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry. Toxins 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Yogendrarajah, P.; Van Poucke, C.; De Meulenaer, B.; De Saeger, S. Development and validation of a QuEChERS based liquid chromatography tandem mass spectrometry method for the determination of multiple mycotoxins in spices. J. Chromatogr. A 2013, 1297, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Fattore, M.; Albrizio, S.; Berrada, H.; Mañes, J. Occurrence of Fusarium mycotoxins and their dietary intake through beer consumption by the European population. Food Chem. 2015, 178, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, J.J.; Cong, J.M.; Cai, Z.X.; Zhang, J.S.; Wang, J.L.; Ren, Y.P. Optimization for quick, easy, cheap, effective, rugged and safe extraction of mycotoxins and veterinary drugs by response surface methodology for application to egg and milk. J. Chromatogr. A 2018, 1532, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; León, N.; Sáez, C.; Martins, C.P.B.; Godula, M.; Yusà, V.; Mañes, J.; Soriano, J.M.; Soler, C. Evaluation of mycotoxins and their metabolites in human breast milk using liquid chromatography coupled to high resolution mass spectrometry. Anal. Chim. Acta 2014, 820, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Scaglioni, P.T.; Becker-Algeri, T.; Drunkler, D.; Badiale-Furlong, E. Aflatoxin B1 and M1 in milk. Anal. Chim. Acta 2014, 829, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Sahin, H.Z.; Celik, M.; Kotay, S.; Kabak, B. Aflatoxins in dairy cow feed, raw milk and milk products from Turkey. Food Addit. Contam. Part B 2016, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Flores-Flores, M.E.; González-Peñas, E. Short communication: Analysis of mycotoxins in Spanish milk. J. Dairy Sci. 2018, 101, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zheng, N.; Wen, F.; Guo, L.; Fu, C.; Ouyang, H.; Lei, S. Multi-mycotoxins analysis in raw milk by ultra high performance liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Food Control 2018, 84, 305–311. [Google Scholar] [CrossRef]
- Tsiplakou, E.; Anagnostopoulos, C.; Liapis, K.; Haroutounian, S.A.; Zervas, G. Determination of mycotoxins in feedstuffs and ruminant’s milk using an easy and simple LC–MS/MS multiresidue method. Talanta 2014, 130, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Flores-Flores, M.E.; González-Peñas, E. An LC–MS/MS method for multi-mycotoxin quantification in cow milk. Food Chem. 2017, 218, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Chu, X.; Ling, Y.; Huang, J.; Chang, J. Multi-mycotoxin analysis in dairy products by liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. J. Chromatogr. A 2014, 1345, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Mañes, J.; Ritieni, A. Urinary levels of enniatin B and its phase I metabolites: First human pilot biomonitoring study. Food Chem. Toxicol. 2018, 118, 454–459. [Google Scholar] [CrossRef] [PubMed]
- European Commission Decision (2002/657/EC) of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and Interpretation of Results. 2002. Available online: https://publications.europa.eu/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en (accessed on 4 April 2018).
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Boudra, H.; Barnouin, J.; Dragacci, S.; Morgavi, D.P. Aflatoxin M1 and ochratoxin A in raw bulk milk from French dairy herds. J. Dairy Sci. 2007, 90, 3197–3201. [Google Scholar] [CrossRef] [PubMed]
- Bilandžić, N.; Varenina, I.; Solomun, B. Aflatoxin M1 in raw milk in Croatia. Food Control 2010, 21, 1279–1281. [Google Scholar] [CrossRef]
- Duarte, S.C.; Almeida, A.M.; Teixeira, A.S.; Pereira, A.L.; Falcão, A.C.; Pena, A.; Lino, C.M. Aflatoxin M1 in marketed milk in Portugal: Assessment of human and animal exposure. Food Control 2013, 30, 411–417. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; Lizarraga, E.; de Cerain, A.L.; González-Peñas, E. Presence of mycotoxins in animal milk: A review. Food Control 2015, 53, 163–176. [Google Scholar] [CrossRef]
- Nassib, T.A.; Guergues, S.N.; Motawee, M.M. Survey for Detection and Determination of Aflatoxins M1 and B1 in local Milk and Certain Dairy Products by Thin Layer Chromatographic Method. Egypt. J. Hosp. Med. 2005, 18, 29–36. [Google Scholar]
- Carvajal, M.; Rojo, F.; Méndez, I.; Bolaños, A. Aflatoxin B1 and its interconverting metabolite aflatoxicol in milk: The situation in Mexico. Food Addit. Contam. 2003, 20, 1077–1086. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Mycotoxins | Retention Time (min) | Elemental Composition | Adduct Ion | Theoretical Mass (m/z) | Measured Mass (m/z) | Accuracy (Δ ppm) | Product Ion (m/z) | Collision Energy (eV) |
|---|---|---|---|---|---|---|---|---|
| AFM1 | 4.60 | C17H12O7 | [M − H]+ | 329.06558 | 329.06511 | −1.43 | 273.07538 229.04909 | 40 |
| AFB1 | 5.02 | C17H12O6 | [M − H]+ | 313.07066 | 313.06958 | −3.45 | 285.07489 269.04373 | 36 |
| Parameters | R2 | SSE (%) | Recovery, % (U′, %; k = 2) | LOD (µg/L) | LOQ (µg/L) | ||
|---|---|---|---|---|---|---|---|
| 0.005 µg/L | 0.01 µg/L | 0.05 µg/L | |||||
| AFM1 | 0.9994 | 65 | 75 (26) | 84 (20) | 91 (16) | 0.001 | 0.002 |
| AF B1 | 0.9996 | 72 | 81 (32) | 87 (24) | 96 (22) | 0.001 | 0.002 |
| Milk Origin | Positive Samples | % Positive Samples | Range (μg/L) | LOQ (μg/L) | References | |
|---|---|---|---|---|---|---|
| Mexico | AFM1 | 117/290 | 1.4 | <LOQ-8.35 | 0.05 | Carvajal et al. [33] |
| AFB1 | 4/290 | 40.3 | <LOQ-0.42 | 0.05 | ||
| Egypt | AFM1 | 11/80 | 13.8 | 0.144–0.378 | n.r. | Nassib et al. [32] |
| AFB1 | 1/80 | 1.2 | <LOQ-3.54 | n.r. | ||
| Brazil | AFM1 | 21/40 | 52.5 | 0.7–1.5 | 0.50 | Scaglioni et al. [18] |
| AFB1 | 7/40 | 17.5 | 0.8–1.7 | 0.25 | ||
| Spain | AFM1 | 0/10 | - | <LOD | 0.025 | Flores-Flores et al. [23] |
| AFB1 | 0/10 | - | <LOD | 0.020 | ||
| Spain | AFM1 | 0/191 | - | <LOD | 0.025 | Flores-Flores et al. [20] |
| AFB1 | 0/191 | - | <LOD | 0.020 | ||
| China | AFM1 | 11/45 | 24.4 | <LOQ-0.03 | 0.01 | Zhou et al. [16] |
| AFB1 | 0/45 | - | <LOD | 0.01 | ||
| China | AFM1 | 247/250 | 98.8 | 0.002–0.028 | 0.001 | Mao et al. [21] |
| AFB1 | 31/250 | 12.4 | <LOQ-0.023 | 0.0033 | ||
| Italy | AFM1 | 0/40 | - | <LOD | 0.002 | This study |
| AFB1 | 0/40 | - | <LOD | 0.002 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Mañes, J.; Ritieni, A. Simultaneous Determination of AFB1 and AFM1 in Milk Samples by Ultra High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry. Beverages 2018, 4, 43. https://doi.org/10.3390/beverages4020043
Rodríguez-Carrasco Y, Izzo L, Gaspari A, Graziani G, Mañes J, Ritieni A. Simultaneous Determination of AFB1 and AFM1 in Milk Samples by Ultra High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry. Beverages. 2018; 4(2):43. https://doi.org/10.3390/beverages4020043
Chicago/Turabian StyleRodríguez-Carrasco, Yelko, Luana Izzo, Anna Gaspari, Giulia Graziani, Jordi Mañes, and Alberto Ritieni. 2018. "Simultaneous Determination of AFB1 and AFM1 in Milk Samples by Ultra High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry" Beverages 4, no. 2: 43. https://doi.org/10.3390/beverages4020043
APA StyleRodríguez-Carrasco, Y., Izzo, L., Gaspari, A., Graziani, G., Mañes, J., & Ritieni, A. (2018). Simultaneous Determination of AFB1 and AFM1 in Milk Samples by Ultra High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry. Beverages, 4(2), 43. https://doi.org/10.3390/beverages4020043