
Microbial Dynamics and Phage Composition Reveal Key Transitions Driving Product Stability in Natural Vinegar Fermentation

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Vinegar Production and Sampling
2.2. Total DNA Extraction
2.3. Viral DNA Extraction
2.4. Library Construction and Shotgun Sequencing
2.5. Bioinformatics and Data Analysis
2.6. KEGG Pathway Annotation
3. Results and Discussion
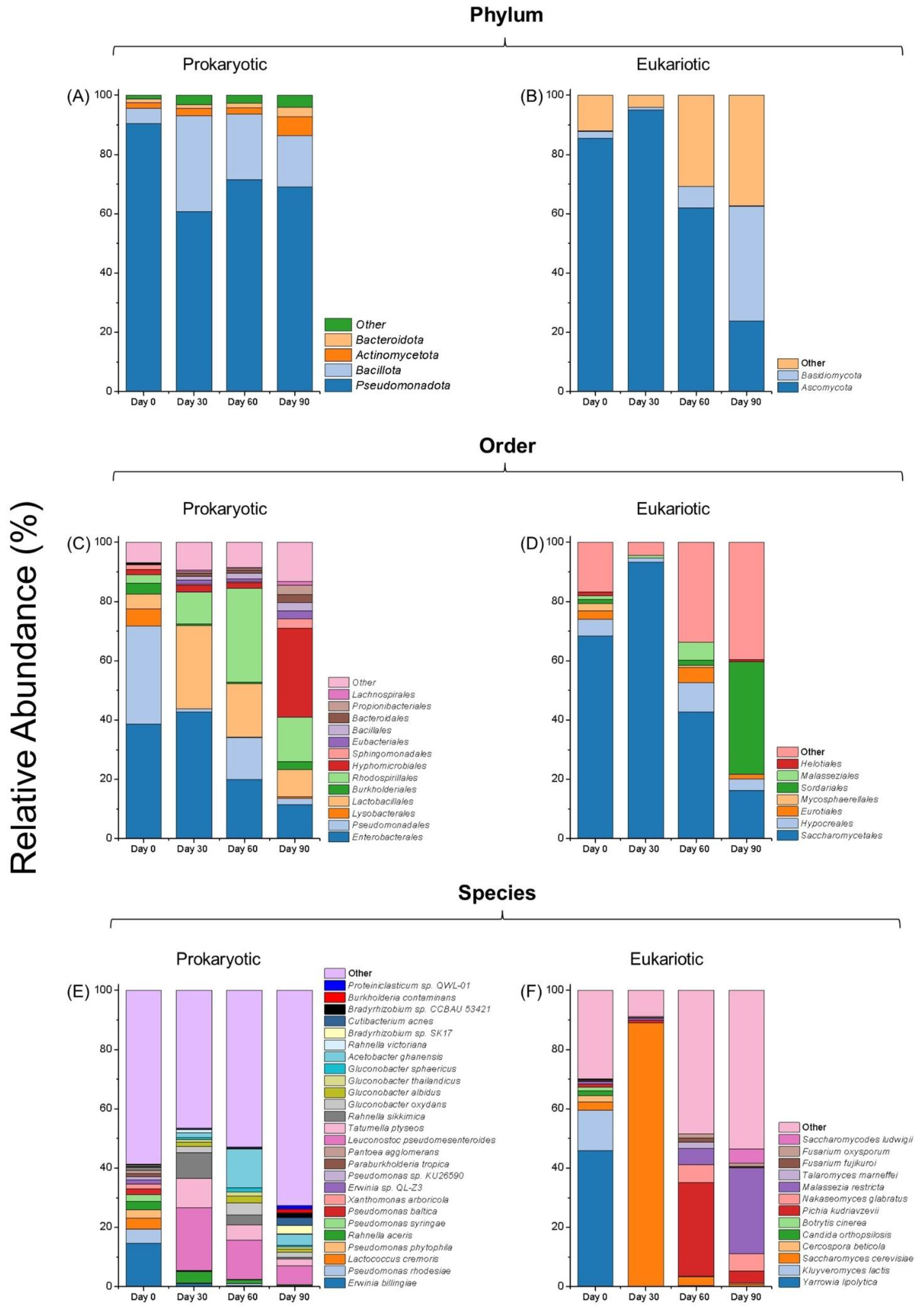
3.1. Dynamic Succession of Bacteria and Fungi
3.1.1. Shotgun Reads
3.1.2. Microbial Diversity
3.1.3. Order-Level Trends
3.1.4. Species-Level Interactions
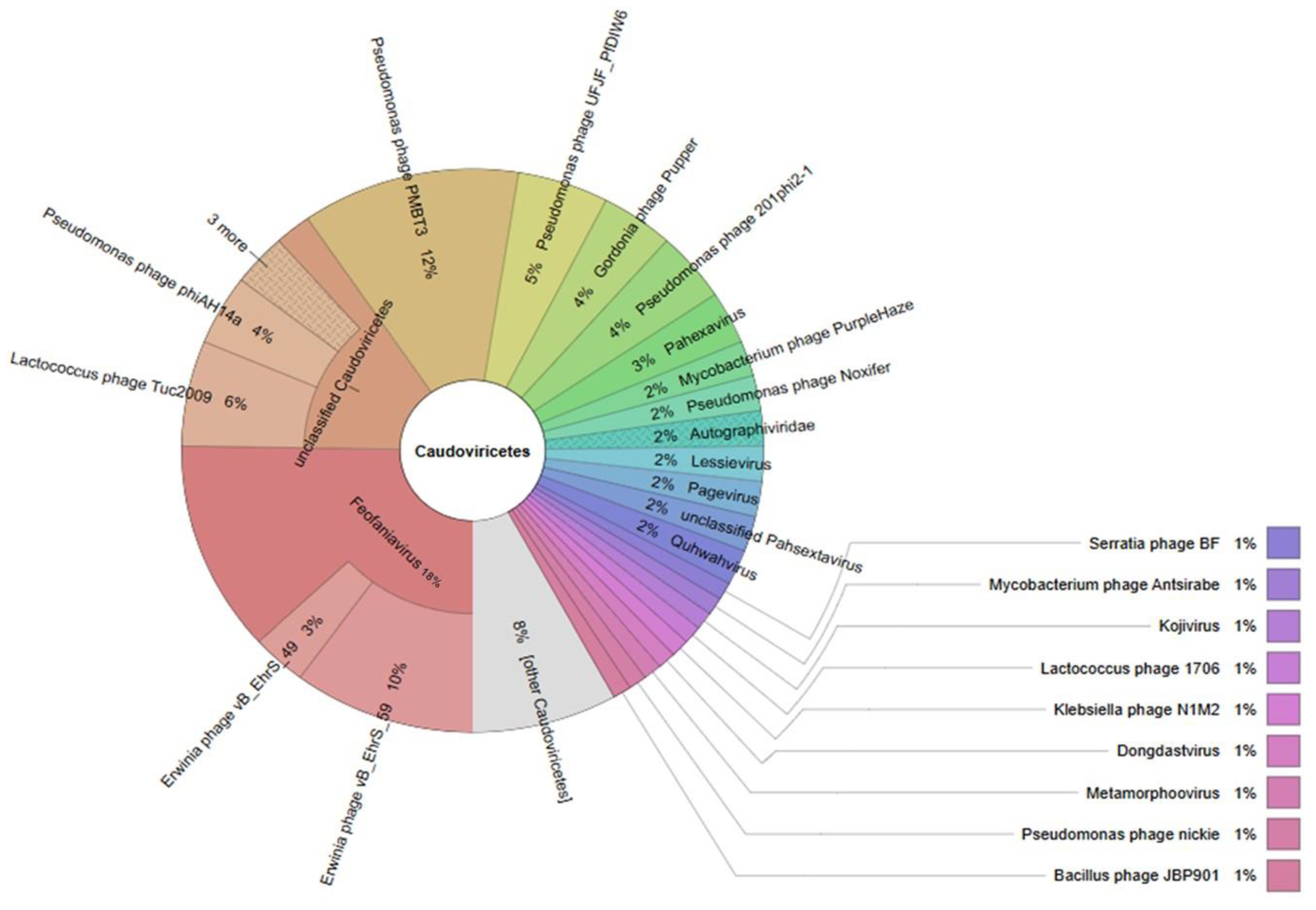
3.2. Bacteriophage Diversity
Predictive Role of Phages in Product Stability
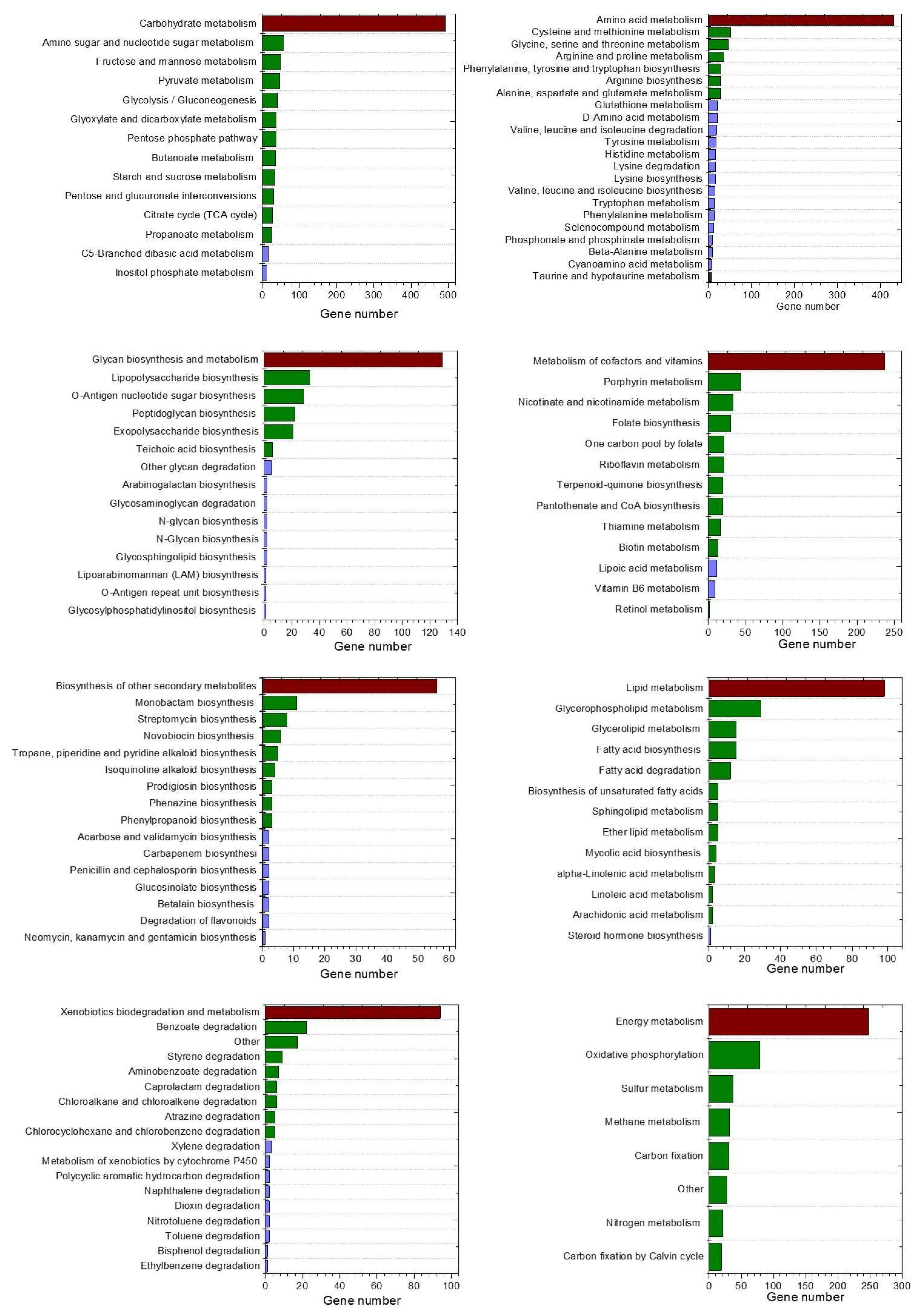
3.3. Predictive Functional Features
3.4. Correlation Between Predominant Species and Predictive Functions
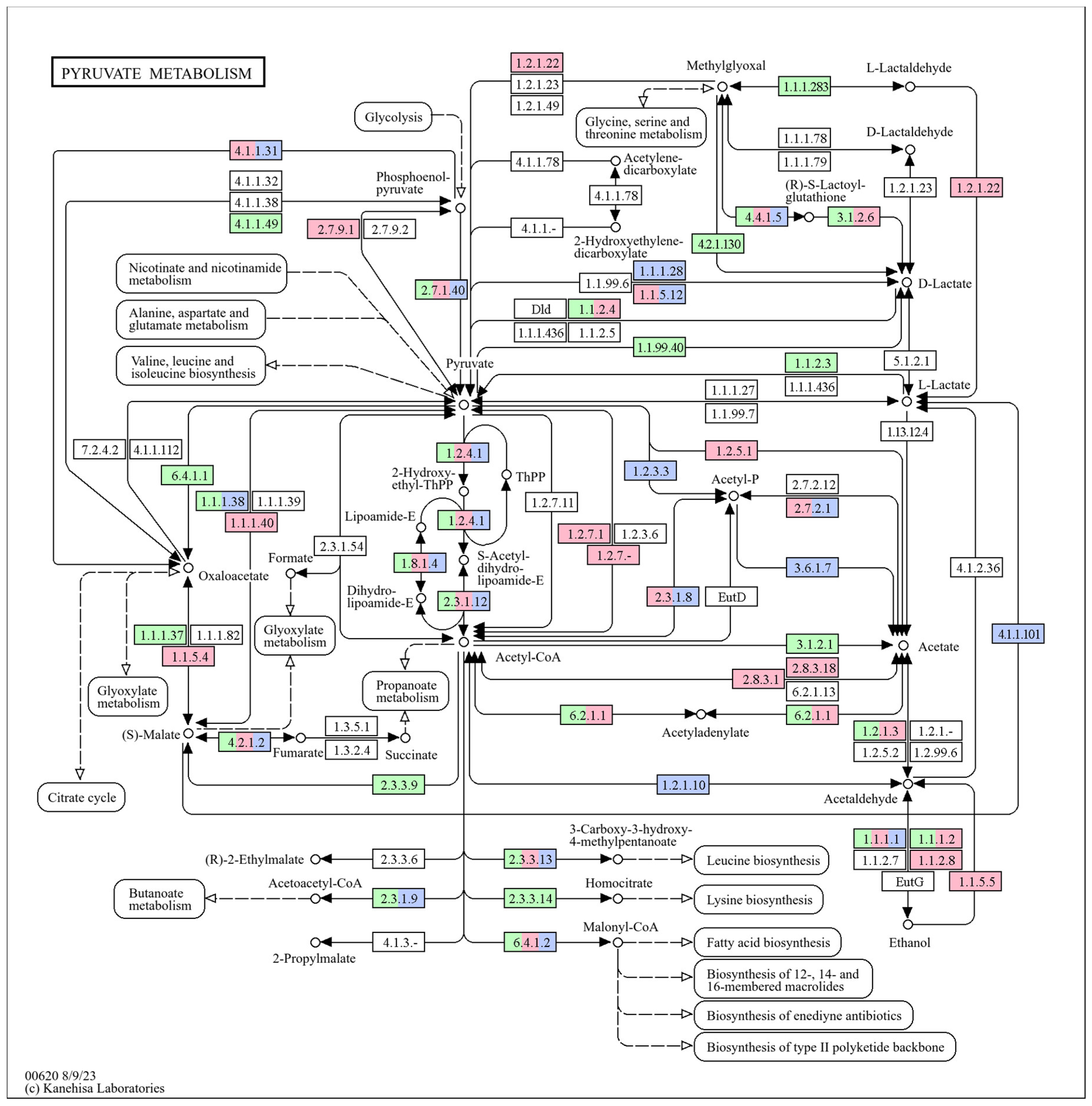
3.5. Pyruvate Metabolism and Pectin Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mas, A.; Troncoso, A.M.; García-Parrilla, M.C.; Torija, M.J. Vinegar; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12384-953-3. [Google Scholar]
- Mota, J.; Vilela, A. Exploring Microbial Dynamics: The Interaction between Yeasts and Acetic Acid Bacteria in Port Wine Vinegar and Its Implications on Chemical Composition and Sensory Acceptance. Fermentation 2024, 10, 421. [Google Scholar] [CrossRef]
- Maske, B.L.; de Mello, A.F.M.; Vale, A.d.S.; Martin, J.G.P.; Soares, D.L.d.O.; Lindner, J.D.D.; Soccol, C.R.; Pereira, G.V. Exploring Diversity and Functional Traits of Lactic Acid Bacteria in Traditional Vinegar Fermentation: A Review. Int. J. Food Microbiol. 2023, 412, 110550. [Google Scholar] [CrossRef]
- Ferrocino, I.; Bellio, A.; Giordano, M.; Macori, G.; Romano, A.; Rantsiou, K.; Decastelli, L.; Cocolin, L. Shotgun Metagenomics and Volatilome Profile of the Microbiota of Fermented Sausages. Appl. Environ. Microbiol. 2018, 84, e02120-17. [Google Scholar] [CrossRef]
- Marco, D.E.; Abram, F. Using Genomics, Metagenomics and Other “Omics” to Assess Valuable Microbial Ecosystem Services and Novel Biotechnological Applications. Front. Microbiol. 2019, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, F.X.; Zeng, Z.; Xu, M.; Sun, F.; Yang, L.; Bi, X.; Lin, Y.; Gao, Y.J.; Hao, H.X.; et al. Advances in Metagenomics and Its Application in Environmental Microorganisms. Front. Microbiol. 2021, 12, 766364. [Google Scholar] [CrossRef] [PubMed]
- Tamang, J.P.; Kharnaior, P.; Pariyar, P.; Thapa, N.; Lar, N.; Win, K.S.; Mar, A.; Nyo, N. Shotgun Sequence-Based Metataxonomic and Predictive Functional Profiles of Pe Poke, a Naturally Fermented Soybean Food of Myanmar. PLoS ONE 2021, 16, e0260777. [Google Scholar] [CrossRef]
- Román-Camacho, J.J.; Mauricio, J.C.; Santos-Dueñas, I.M.; García-Martínez, T.; García-García, I. Recent Advances in Applying Omic Technologies for Studying Acetic Acid Bacteria in Industrial Vinegar Production: A Comprehensive Review. Biotechnol. J. 2024, 19, e2300566. [Google Scholar] [CrossRef]
- Weiland-Bräuer, N. Friends or Foes—Microbial Interactions in Nature. Biology 2021, 10, 496. [Google Scholar] [CrossRef]
- Chin, Y.W.; Hong, S.P.; Lim, S.D.; Yi, S.H. Investigation of Microbial Community of Korean Soy Sauce (Ganjang) Using Shotgun Metagenomic Sequencing and Its Relationship with Sensory Characteristics. Microorganisms 2024, 12, 2559. [Google Scholar] [CrossRef]
- Ranveer, S.A.; Dasriya, V.; Ahmad, M.F.; Dhillon, H.S.; Samtiya, M.; Shama, E.; Anand, T.; Dhewa, T.; Chaudhary, V.; Chaudhary, P.; et al. Positive and Negative Aspects of Bacteriophages and Their Immense Role in the Food Chain. njp Sci. Food 2024, 8, 1. [Google Scholar] [CrossRef]
- Ma, J.; Qian, C.; Hu, Q.; Zhang, J.; Gu, G.; Liang, X.; Zhang, L. The Bacteriome-Coupled Phage Communities Continuously Contract and Shift to Orchestrate the Traditional Rice Vinegar Fermentation. Food Res. Int. 2024, 184, 114244. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Fujibuchi, W.; Kanehisa, M. Computation with the KEGG Pathway Database. BioSystems 1998, 47, 119–128. [Google Scholar] [CrossRef]
- Maske, B.L.; Ruiz, I.; Vale, A.d.S.; Sampaio, V.d.M.; El Kadri, N.K.; Soccol, C.R.; Pereira, G.V. Predicting the Microbiome and Metabolome Dynamics of Natural Apple Fermentation Towards the Development of Enhanced Functional Vinegar. Fermentation 2024, 10, 552. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Edwin, N.R.; Fitzpatrick, A.H.; Brennan, F.; Abram, F.; O’Sullivan, O. An In-Depth Evaluation of Metagenomic Classifiers for Soil Microbiomes. Environ. Microbiome 2024, 19, 19. [Google Scholar] [CrossRef]
- Pusadkar, V.; Azad, R.K. Benchmarking Metagenomic Classifiers on Simulated Ancient and Modern Metagenomic Data. Microorganisms 2023, 11, 2478. [Google Scholar] [CrossRef]
- LaBauve, A.E.; Wargo, M.J. Growth and Laboratory Maintenance of Pseudomonas Aeruginosa. Curr. Protoc. Microbiol. 2012, 25, 6E.1.1–6E.1.8. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.P. Advances in Microbiological Quality Control; Woodhead Publishing Limited: Cambridge, UK, 2010; ISBN 978-1-84569-484-5. [Google Scholar]
- Qiu, X.; Zhang, Y.; Hong, H. Classification of Acetic Acid Bacteria and Their Acid Resistant Mechanism. AMB Express 2021, 11, 29. [Google Scholar] [CrossRef]
- Niyomvong, N.; Sritawan, R.; Keabpimai, J.; Trakunjae, C.; Boondaeng, A. Comparison of the Chemical Properties of Vinegar Obtained via One-Step Fermentation and Sequential Fermentation from Dragon Fruit and Pineapple. Beverages 2022, 8, 74. [Google Scholar] [CrossRef]
- Erasmus, D.J.; Van Der Merwe, G.K.; Van Vuuren, H.J.J. Genome-Wide Expression Analyses: Metabolic Adaptation of Saccharomyces Cerevisiae to High Sugar Stress. FEMS Yeast Res. 2003, 3, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Dashko, S.; Zhou, N.; Compagno, C.; Piškur, J. Why, When, and How Did Yeast Evolve Alcoholic Fermentation? FEMS Yeast Res. 2014, 14, 826–832. [Google Scholar] [CrossRef]
- Karanth, S.; Feng, S.; Patra, D.; Pradhan, A.K. Linking Microbial Contamination to Food Spoilage and Food Waste: The Role of Smart Packaging, Spoilage Risk Assessments, and Date Labeling. Front. Microbiol. 2023, 14, 1198124. [Google Scholar] [CrossRef]
- De Oliveira Junqueira, A.C.; de Melo Pereira, G.V.; Coral Medina, J.D.; Alvear, M.C.R.; Rosero, R.; de Carvalho Neto, D.P.; Enríquez, H.G.; Soccol, C.R. First Description of Bacterial and Fungal Communities in Colombian Coffee Beans Fermentation Analysed Using Illumina-Based Amplicon Sequencing. Sci. Rep. 2019, 9, 8794. [Google Scholar] [CrossRef]
- Fleet, G.H. Yeast Interactions and Wine Flavour. Int. J. Food Microbiol. 2003, 86, 11–22. [Google Scholar] [CrossRef]
- Atasoy, M.; Ordóñez, A.Á.; Cenian, A.; Djukić-Vuković, A.; Lund, P.A.; Ozogul, F.; Trček, J.; Ziv, C.; De Biase, D. Exploitation of Microbial Activities at Low PH to Enhance Planetary Health. FEMS Microbiol. Rev. 2024, 48, fuad062. [Google Scholar] [CrossRef]
- Nie, Z.; Zheng, Y.; Wang, M.; Han, Y.; Wang, Y.; Luo, J.; Niu, D. Exploring Microbial Succession and Diversity during Solid-State Fermentation of Tianjin Duliu Mature Vinegar. Bioresour. Technol. 2013, 148, 325–333. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, P.; Zhou, X.; Zheng, J.; Ma, Y.; Liu, C.; Wu, T.; Li, H.; Wang, X.; Wang, H.; et al. Isolation, Identification, and Characterization of an Acid-Tolerant Pichia Kudriavzevii and Exploration of Its Acetic Acid Tolerance Mechanism. Fermentation 2023, 9, 540. [Google Scholar] [CrossRef]
- Pereira, G.V.M.; Alvarez, J.P.; Neto, D.P.d.C.; Soccol, V.T.; Tanobe, V.O.A.; Rogez, H.; Góes-Neto, A.; Soccol, C.R. Great Intraspecies Diversity of Pichia Kudriavzevii in Cocoa Fermentation Highlights the Importance of Yeast Strain Selection for Flavor Modulation of Cocoa Beans. LWT 2017, 84, 290–297. [Google Scholar] [CrossRef]
- Vijaya Chandra, S.H.; Srinivas, R.; Dawson, T.L.; Common, J.E. Cutaneous Malassezia: Commensal, Pathogen, or Protector? Front. Cell. Infect. Microbiol. 2021, 10, 614446. [Google Scholar] [CrossRef]
- Naureen, Z.; Dautaj, A.; Anpilogov, K.; Camilleri, G.; Dhuli, K.; Tanzi, B.; Maltese, P.E.; Cristofoli, F.; De Antoni, L.; Beccari, T.; et al. Bacteriophages Presence in Nature and Their Role in the Natural Selection of Bacterial Populations. Acta Biomed. 2020, 91, e2020024. [Google Scholar] [CrossRef]
- Warriner, K.; Namvar, A. Biosensors for Foodborne Pathogen Detection, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 4, ISBN 978-0-44464-047-5. [Google Scholar]
- Hylling, O.; Carstens, A.B.; Kot, W.; Hansen, M.; Neve, H.; Franz, C.M.A.P.; Johansen, A.; Ellegaard-Jensen, L.; Hansen, L.H. Two Novel Bacteriophage Genera from a Groundwater Reservoir Highlight Subsurface Environments as Underexplored Biotopes in Bacteriophage Ecology. Sci. Rep. 2020, 10, 11879. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hussain, W.; Chen, Y.; Xu, P.; Yang, X.; Wang, H.; Zhang, X.; Fu, Q.; Wang, S. A New Type of Pseudomonas Aeruginosa Phage with Potential as a Natural Food Additive for Eradicating Biofilms and Combating Multidrug-Resistant Strains. Food Control 2025, 168, 110888. [Google Scholar] [CrossRef]
- Hungaro, H.M.; Vidigal, P.M.P.; Do Nascimento, E.C.; Gomes da Costa Oliveira, F.; Gontijo, M.T.P.; Lopez, M.E.S. Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas Fluorescens-Phage with Potential for Biocontrol in the Dairy Industry. Viruses 2022, 14, 629. [Google Scholar] [CrossRef]
- Do Nascimento, E.C.; Sabino, M.C.; Corguinha, L.d.R.; Targino, B.N.; Lange, C.C.; Pinto, C.L.d.O.; Pinto, P.d.F.; Vidigal, P.M.P.; Sant’Ana, A.S.; Hungaro, H.M. Lytic Bacteriophages UFJF_PfDIW6 and UFJF_PfSW6 Prevent Pseudomonas Fluorescens Growth in Vitro and the Proteolytic-Caused Spoilage of Raw Milk during Chilled Storage. Food Microbiol. 2022, 101, 103892. [Google Scholar] [CrossRef]
- Thomas, J.A.; Rolando, M.R.; Carroll, C.A.; Shen, P.S.; Belnap, D.M.; Weintraub, S.T.; Serwer, P.; Hardies, S.C. Characterization of Pseudomonas Chlororaphis Myovirus 201φ2-1 via Genomic Sequencing, Mass Spectrometry, and Electron Microscopy. Virology 2008, 376, 330–338. [Google Scholar] [CrossRef]
- Morozova, V.; Kozlova, Y.; Tikunov, A.; Babkin, I.; Ushakova, T.; Bardasheva, A.; Jdeed, G.; Zhirakovskaya, E.; Mogileva, A.; Netesov, S.; et al. Identification, Characterization, and Genome Analysis of Two Novel Temperate Pseudomonas Protegens Phages PseuP_222 and PseuP_224. Microorganisms 2023, 11, 1456. [Google Scholar] [CrossRef]
- Kwon, J.; Kim, S.W.; Kim, S.G.; Kang, J.W.; Jung, W.J.; Bin Lee, S.; Lee, Y.M.; Giri, S.S.; Chi, C.; Park, S.C. The Characterization of a Novel Phage, Ppa_snuabm_dt01, Infecting Pseudomonas aeruginosa. Microorganisms 2021, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Martino, G.; Holtappels, D.; Vallino, M.; Chiapello, M.; Turina, M.; Lavigne, R.; Wagemans, J.; Ciuffo, M. Molecular Characterization and Taxonomic Assignment of Three Phage Isolates from a Collection Infecting Pseudomonas Syringae Pv. Actinidiae and p. Syringae Pv. Phaseolicola from Northern Italy. Viruses 2021, 13, 2083. [Google Scholar] [CrossRef] [PubMed]
- Zlatohurska, M.; Gorb, T.; Romaniuk, L.; Korol, N.; Faidiuk, Y.; Kropinski, A.M.; Kushkina, A.; Tovkach, F. Complete Genome Sequence Analysis of Temperate Erwinia Bacteriophages 49 and 59. J. Basic Microbiol. 2019, 59, 754–764. [Google Scholar] [CrossRef]
- Li, X.; Ding, W.; Li, Z.; Yan, Y.; Tong, Y.; Xu, J.; Li, M. VB_CacS-HV1 as a Novel Pahexavirus Bacteriophage with Lytic and Anti-Biofilm Potential against Cutibacterium Acnes. Microorganisms 2024, 12, 1566. [Google Scholar] [CrossRef]
- Mayslich, C.; Grange, P.A.; Dupin, N. Cutibacterium acnes as an Opportunistic Pathogen: An Update of Its Virulence-Associated Factors. Microorganisms 2021, 9, 303. [Google Scholar] [CrossRef]
- Sinha, A.K.; Løbner-Olesen, A.; Riber, L. Bacterial Chromosome Replication and DNA Repair During the Stringent Response. Front. Microbiol. 2020, 11, 582113. [Google Scholar] [CrossRef]
- Wei, J.; Lu, J.; Nie, Y.; Li, C.; Du, H.; Xu, Y. Amino Acids Drive the Deterministic Assembly Process of Fungal Community and Affect the Flavor Metabolites in Baijiu Fermentation. Microbiol. Spectr. 2023, 11, e0264022. [Google Scholar] [CrossRef]
- Jeckelmann, J.M.; Erni, B. Transporters of Glucose and Other Carbohydrates in Bacteria. Pflugers Arch. Eur. J. Physiol. 2020, 472, 1129–1153. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Liu, C.J.; Zeng, X.Q.; Zhang, H.Y.; Luo, Y.Y.; Li, X.R. Metagenomic and Metatranscriptomic Analysis of the Microbial Community Structure and Metabolic Potential of Fermented Soybean in Yunnan Province. Food Sci. Technol. 2020, 40, 18–25. [Google Scholar] [CrossRef]
- Pelicaen, R.; Gonze, D.; Teusink, B.; De Vuyst, L.; Weckx, S. Genome-Scale Metabolic Reconstruction of Acetobacter pasteurianus 386B, a Candidate Functional Starter Culture for Cocoa Bean Fermentation. Front. Microbiol. 2019, 10, 2801. [Google Scholar] [CrossRef]
- Kondakova, T.; D’Heygère, F.; Feuilloley, M.J.; Orange, N.; Heipieper, H.J.; Duclairoir Poc, C. Glycerophospholipid Synthesis and Functions in Pseudomonas. Chem. Phys. Lipids 2015, 190, 27–42. [Google Scholar] [CrossRef]
- Es-Sbata, I.; Castro-Mejías, R.; Rodríguez-Dodero, C.; Zouhair, R.; Durán-Guerrero, E. Sensory Analysis as a Simple and Low-Cost Tool to Evaluate and Valorize a New Product from Local Fruits in Rural Communities: The Case of Highly Aromatic Vinegar from Prickly Pear Fruits. Beverages 2023, 9, 74. [Google Scholar] [CrossRef]
- Dzialo, M.C.; Park, R.; Steensels, J.; Lievens, B.; Verstrepen, K.J. Physiology, Ecology and Industrial Applications of Aroma Formation in Yeast. FEMS Microbiol. Rev. 2017, 41, S95–S128. [Google Scholar] [CrossRef]
- Hua, S.; Wang, Y.; Wang, L.; Zhou, Q.; Li, Z.; Liu, P.; Wang, K.; Zhu, Y.; Han, D.; Yu, Y. Regulatory Mechanisms of Acetic Acid, Ethanol and High Temperature Tolerances of Acetic Acid Bacteria during Vinegar Production. Microb. Cell Factories 2024, 23, 324. [Google Scholar] [CrossRef] [PubMed]
- Dols, M.; Chraibi, W.; Remaud-Simeon, M.; Lindley, N.D.; Monsan, P.F. Growth and Energetics of Leuconostoc Mesenteroides NRRL B-1299 during Metabolism of Various Sugars and Their Consequences for Dextransucrase Production. Appl. Environ. Microbiol. 1997, 63, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Meng, Z.; Ma, X.; Zhao, S.; An, Y.; Xiao, Z. Characterization and Regulation of the Acetolactate Synthase Genes Involved in Acetoin Biosynthesis in Acetobacter Pasteurianus. Foods 2021, 10, 1013. [Google Scholar] [CrossRef]
- Dringen, R.; Hoepken, H.H.; Minich, T.; Ruedig, C. Pentose Phosphate Pathway and NADPH Metabolism. In Handbook of Neurochemistry and Molecular Neurobiology; Springer: New York, NY, USA, 2007; pp. 41–62. [Google Scholar] [CrossRef]
- Muslu Can, A.; Metin Yildirim, R.; Karadag, A. The Properties of Pectin Extracted from the Residues of Vinegar-Fermented Apple and Apple Pomace. Fermentation 2024, 10, 556. [Google Scholar] [CrossRef]
- Özcan, E.; Selvi, S.S.; Nikerel, E.; Teusink, B.; Toksoy Öner, E.; Çakır, T. A Genome-Scale Metabolic Network of the Aroma Bacterium Leuconostoc Mesenteroides Subsp. Cremoris. Appl. Microbiol. Biotechnol. 2019, 103, 3153–3165. [Google Scholar] [CrossRef]
- Sprotte, S.; Brinks, E.; Wagner, N.; Kropinski, A.M.; Neve, H.; Franz, C.M.A.P. Characterization of the First Pseudomonas Grimontii Bacteriophage, PMBT3. Arch. Virol. 2021, 166, 2887–2894. [Google Scholar] [CrossRef]
- Abdollahzadeh, R.; Pazhang, M.; Najavand, S.; Fallahzadeh-Mamaghani, V.; Amani-Ghadim, A.R. Screening of Pectinase-Producing Bacteria from Farmlands and Optimization of Enzyme Production from Selected Strain by RSM. Folia Microbiol. 2020, 65, 705–719. [Google Scholar] [CrossRef]
- Lv, L.; Luo, J.; Ahmed, T.; Zaki, H.E.M.; Tian, Y.; Shahid, M.S.; Chen, J.; Li, B. Beneficial Effect and Potential Risk of Pantoea on Rice Production. Plants 2022, 11, 2608. [Google Scholar] [CrossRef] [PubMed]
- Vale, A.d.S.; Pereira, C.M.T.; De Dea Lindner, J.; Rodrigues, L.R.S.; El Kadri, N.K.; Pagnoncelli, M.G.B.; Kaur Brar, S.; Soccol, C.R.; Pereira, G.V.d.M. Exploring Microbial Influence on Flavor Development during Coffee Processing in Humid Subtropical Climate through Metagenetic–Metabolomics Analysis. Foods 2024, 13, 1871. [Google Scholar] [CrossRef] [PubMed]
- Manyapu, V.; Lepcha, A.; Sharma, S.K.; Kumar, R. Role of Psychrotrophic Bacteria and Cold-Active Enzymes in Composting Methods Adopted in Cold Regions. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 2022; pp. 1–26. [Google Scholar]
- Mladenović, K.G.; Grujović, M.Ž.; Kiš, S.F.M.; Tkalec, V.J.; Stefanović, O.D.; Kocić-Tanackov, S.D. Enterobacteriaceae in Food Safety with an Emphasis on Raw Milk and Meat. Appl. Microbiol. Biotechnol. 2021, 105, 8615–8627. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway/Metabolite * | Acetobacter ghanensis | Leuconostoc pseudomesenteroides | Saccharomyces cerevisiae |
|---|---|---|---|
| Amino Acids | |||
| Alanine | High ** | Moderate | High |
| Valine | High | Moderate | None |
| Leucine | High | Moderate | None |
| Isoleucine | Moderate | Moderate | None |
| Glutamate | Moderate | None | High |
| Proline | None | None | High |
| Lysine | None | None | Moderate |
| Phenylalanine | None | High | None |
| Tyrosine | None | High | None |
| Tryptophan | None | High | None |
| Methionine | Moderate | None | Moderate |
| Cysteine | Moderate | None | Moderate |
| Pyruvate Metabolism | |||
| Acetyl-CoA Production | High | Moderate | High |
| Lactate Production | None | High | Moderate |
| Ethanol Production | None | None | High |
| Acetate Production | High | None | None |
| Oxaloacetate Formation | Moderate | None | Moderate |
| Formate Production | Moderate | None | None |
| Succinate Production | Moderate | None | None |
| Leucine/Isoleucine Synthesis | High | Moderate | None |
| Fructose and Mannose | |||
| Fructose Utilization | High | Moderate | High |
| Mannose Utilization | High | High | Moderate |
| D-Mannitol Production | High | High | None |
| Sorbitol Pathway | Moderate | High | None |
| Glycolysis Linkage | High | Moderate | High |
| L-Fucose Utilization | None | Moderate | None |
| L-Rhamnose Utilization | None | None | Moderate |
| Fructose-6P Conversion | High | High | High |
| Starch and Sucrose | |||
| Sucrose Utilization | High | High | High |
| Starch Degradation | Moderate | None | High |
| Maltose Utilization | None | None | High |
| Trehalose Metabolism | High | Moderate | High |
| Levan Biosynthesis | High | High | None |
| Inulin Utilization | Moderate | None | None |
| Cellobiose Utilization | High | None | None |
| Glucose-6P Conversion | High | High | High |
| Secondary Metabolism | |||
| Phenolic Compound Synthesis | High | Moderate | High |
| Terpenoid Biosynthesis | None | None | Moderate |
| Polyketide Biosynthesis | None | None | High |
| Alkaloid Synthesis | None | None | None |
| Non-ribosomal Peptide Biosynthesis | Moderate | None | High |
| Flavonoid Synthesis | High | Moderate | None |
| Isoprenoid Biosynthesis | Moderate | None | High |
| Steroid Biosynthesis | None | None | High |
| Pentose Phosphate Pathway | |||
| Glucose-6P Dehydrogenase | High | Moderate | High |
| Ribulose-5P Epimerase | Moderate | High | High |
| Transketolase Activity | High | High | High |
| D-Ribose Synthesis | High | Moderate | High |
| Sedoheptulose-7P Synthesis | High | High | High |
| Nucleotide Sugar Biosynthesis | Moderate | Moderate | High |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Melo Pereira, G.V.; Maske, B.L.; da Silva Vale, A.; de Carvalho, J.C.; Pagnoncelli, M.G.B.; Soccol, C.R. Microbial Dynamics and Phage Composition Reveal Key Transitions Driving Product Stability in Natural Vinegar Fermentation. Beverages 2025, 11, 71. https://doi.org/10.3390/beverages11030071
de Melo Pereira GV, Maske BL, da Silva Vale A, de Carvalho JC, Pagnoncelli MGB, Soccol CR. Microbial Dynamics and Phage Composition Reveal Key Transitions Driving Product Stability in Natural Vinegar Fermentation. Beverages. 2025; 11(3):71. https://doi.org/10.3390/beverages11030071
Chicago/Turabian Stylede Melo Pereira, Gilberto Vinícius, Bruna Leal Maske, Alexander da Silva Vale, Júlio César de Carvalho, Maria Giovana Binder Pagnoncelli, and Carlos Ricardo Soccol. 2025. "Microbial Dynamics and Phage Composition Reveal Key Transitions Driving Product Stability in Natural Vinegar Fermentation" Beverages 11, no. 3: 71. https://doi.org/10.3390/beverages11030071
APA Stylede Melo Pereira, G. V., Maske, B. L., da Silva Vale, A., de Carvalho, J. C., Pagnoncelli, M. G. B., & Soccol, C. R. (2025). Microbial Dynamics and Phage Composition Reveal Key Transitions Driving Product Stability in Natural Vinegar Fermentation. Beverages, 11(3), 71. https://doi.org/10.3390/beverages11030071