
DropDAE: Denosing Autoencoder with Contrastive Learning for Addressing Dropout Events in scRNA-seq Data




Abstract
1. Introduction
2. Methods
2.1. Overview of DropDAE Method
- Corrupt the input scRNA-seq expression matrix by manually adding dropout noise.
- Pass the corrupted input through the encoder of denoising autoencoder to obtain the latent representation (bottleneck) z.
- Perform a clustering method (e.g., K-means or consensus clustering) on the latent representation z to generate pseudo-labels.
- Sample anchor–positive–negative triplets from the latent space based on the pseudo-labels. Compute the triplet loss to encourage separation.
- Pass the bottleneck z to the decoder of the denosing autoencoder to obtain the reconstructed output. Compute the mean squared error (MSE) between the reconstructed and original input.
- Compute the total loss as the weighted sum of MSE and triplet loss. Update the model parameters via backpropagation.
2.2. Clustering on the Bottleneck Latent Representations: K-Means and Consensus Clustering
2.3. Simulation Settings
2.4. Implementation of Competing Methods
2.5. Evaluation of Downstream Analyses
3. Results
3.1. DropDAE Restores and Improves the Original Clustering Pattern
3.2. Dropdae Exhibits Robustness Under Various Scenarios
3.3. DropDAE Helps Cell Type Identification in the PBMC Dataset
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Gong, W.; Kwak, I.Y.; Pota, P.; Koyano-Nakagawa, N.; Garry, D.J. DrImpute: Imputing dropout events in single cell RNA sequencing data. BMC Bioinform. 2018, 19, 220. [Google Scholar] [CrossRef]
- Juan, W.; Ahn, K.W.; Chen, Y.G.; Lin, C.W. CCI: A Consensus Clustering-Based Imputation Method for Addressing Dropout Events in scRNA-Seq Data. Bioengineering 2025, 12, 31. [Google Scholar] [CrossRef]
- Tracy, S.; Yuan, G.C.; Dries, R. RESCUE: Imputing dropout events in single-cell RNA-sequencing data. BMC Bioinform. 2019, 20, 388. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Li, W.V.; Li, J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Condon, A.; Shah, S.P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 2018, 9, 2002. [Google Scholar] [CrossRef]
- Tian, T.; Wan, J.; Song, Q.; Wei, Z. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat. Mach. Intell. 2019, 1, 191–198. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, M.; Yang, B.; Tong, T.; Zhang, B.; Tang, N. scDLC: A deep learning framework to classify large sample single-cell RNA-seq data. BMC Genom. 2022, 23, 504. [Google Scholar] [CrossRef]
- Vincent, P.; Larochelle, H.; Lajoie, I.; Bengio, Y.; Manzagol, P.A.; Bottou, L. Stacked denoising autoencoders: Learning useful representations in a deep network with a local denoising criterion. J. Mach. Learn. Res. 2010, 11, 3371–3408. [Google Scholar]
- Hadsell, R.; Chopra, S.; LeCun, Y. Dimensionality reduction by learning an invariant mapping. In Proceedings of the 2006 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’06), New York, NY, USA, 17–22 June 2006; Volume 2, pp. 1735–1742. [Google Scholar]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 174. [Google Scholar] [CrossRef]
- Xi, N.M.; Li, J.J. Exploring the optimization of autoencoder design for imputing single-cell RNA sequencing data. Comput. Struct. Biotechnol. J. 2023, 21, 4079–4095. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Tapaswi, M.; Sarfraz, M.S.; Stiefelhagen, R. Clustering based contrastive learning for improving face representations. In Proceedings of the 2020 15th IEEE International Conference on Automatic Face and Gesture Recognition (FG 2020), Buenos Aires, Argentina, 16–20 November 2020; pp. 109–116. [Google Scholar]
- Li, Y.; Hu, P.; Liu, Z.; Peng, D.; Zhou, J.T.; Peng, X. Contrastive clustering. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtual Event, 2–9 February 2021; Volume 35, pp. 8547–8555. [Google Scholar]
- Schroff, F.; Kalenichenko, D.; Philbin, J. FaceNet: A Unified Embedding for Face Recognition and Clustering. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Zhu, X.; Zhang, J.; Xu, Y.; Wang, J.; Peng, X.; Li, H.D. Single-cell clustering based on shared nearest neighbor and graph partitioning. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Rand, W.M. Objective criteria for the evaluation of clustering methods. J. Am. Stat. Assoc. 1971, 66, 846–850. [Google Scholar] [CrossRef]
- Kumari, S.; Maurya, S.; Goyal, P.; Balasubramaniam, S.S.; Goyal, N. Scalable parallel algorithms for shared nearest neighbor clustering. In Proceedings of the 2016 IEEE 23rd International Conference on High Performance Computing (HiPC), Hyderabad, India, 19–22 December 2016; pp. 72–81. [Google Scholar]
- Halkidi, M.; Batistakis, Y.; Vazirgiannis, M. Cluster validity methods: Part I. ACM Sigmod Rec. 2002, 31, 40–45. [Google Scholar] [CrossRef]
- Vargo, A.H.; Gilbert, A.C. A rank-based marker selection method for high throughput scRNA-seq data. BMC Bioinform. 2020, 21, 477. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or biology: The zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Vincent, P.; Larochelle, H.; Bengio, Y.; Manzagol, P.A. Extracting and composing robust features with denoising autoencoders. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; pp. 1096–1103. [Google Scholar]
- Samek, W.; Wiegand, T.; Müller, K.R. Explainable artificial intelligence: Understanding, visualizing and interpreting deep learning models. arXiv 2017, arXiv:1708.08296. [Google Scholar]

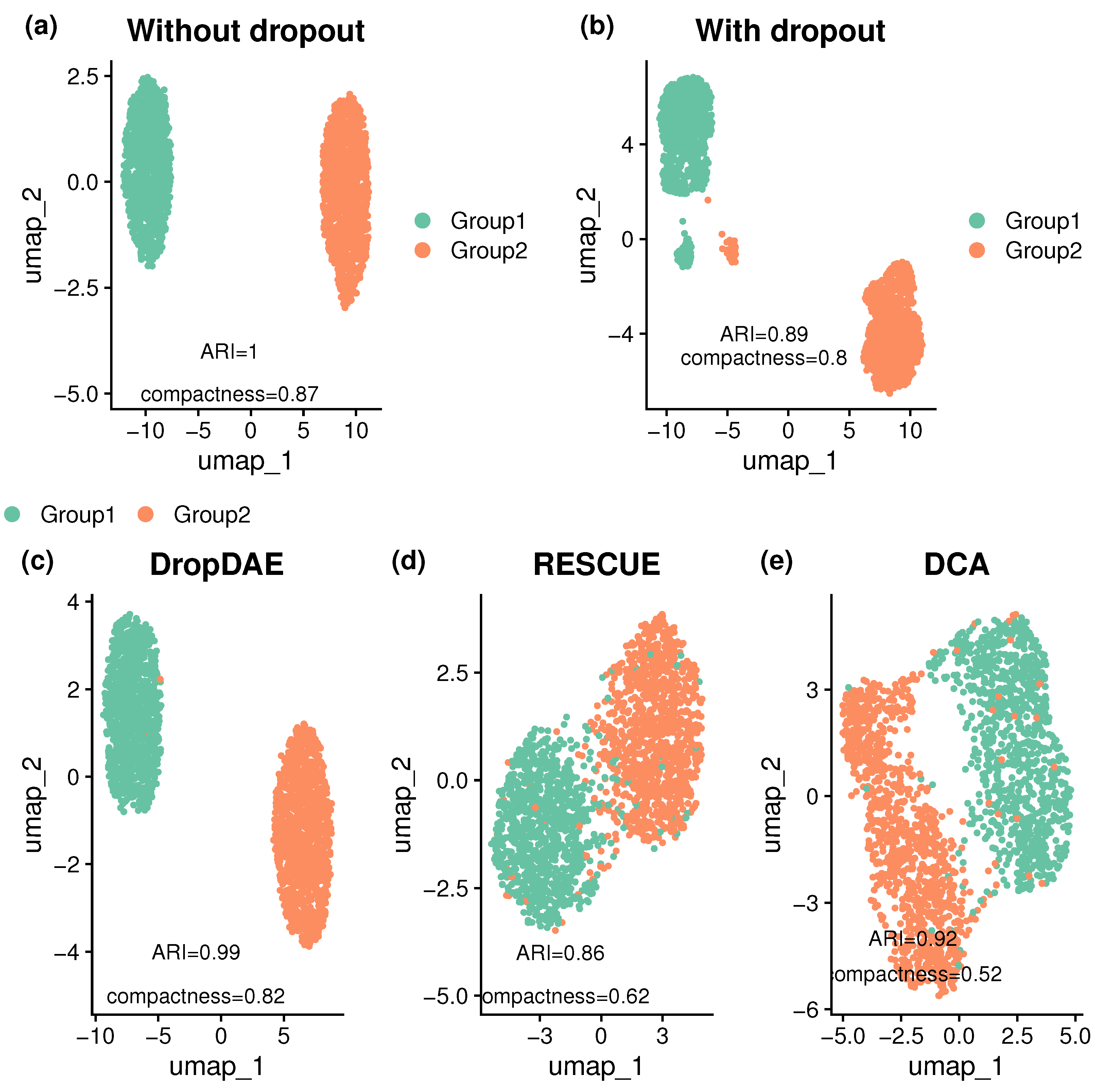

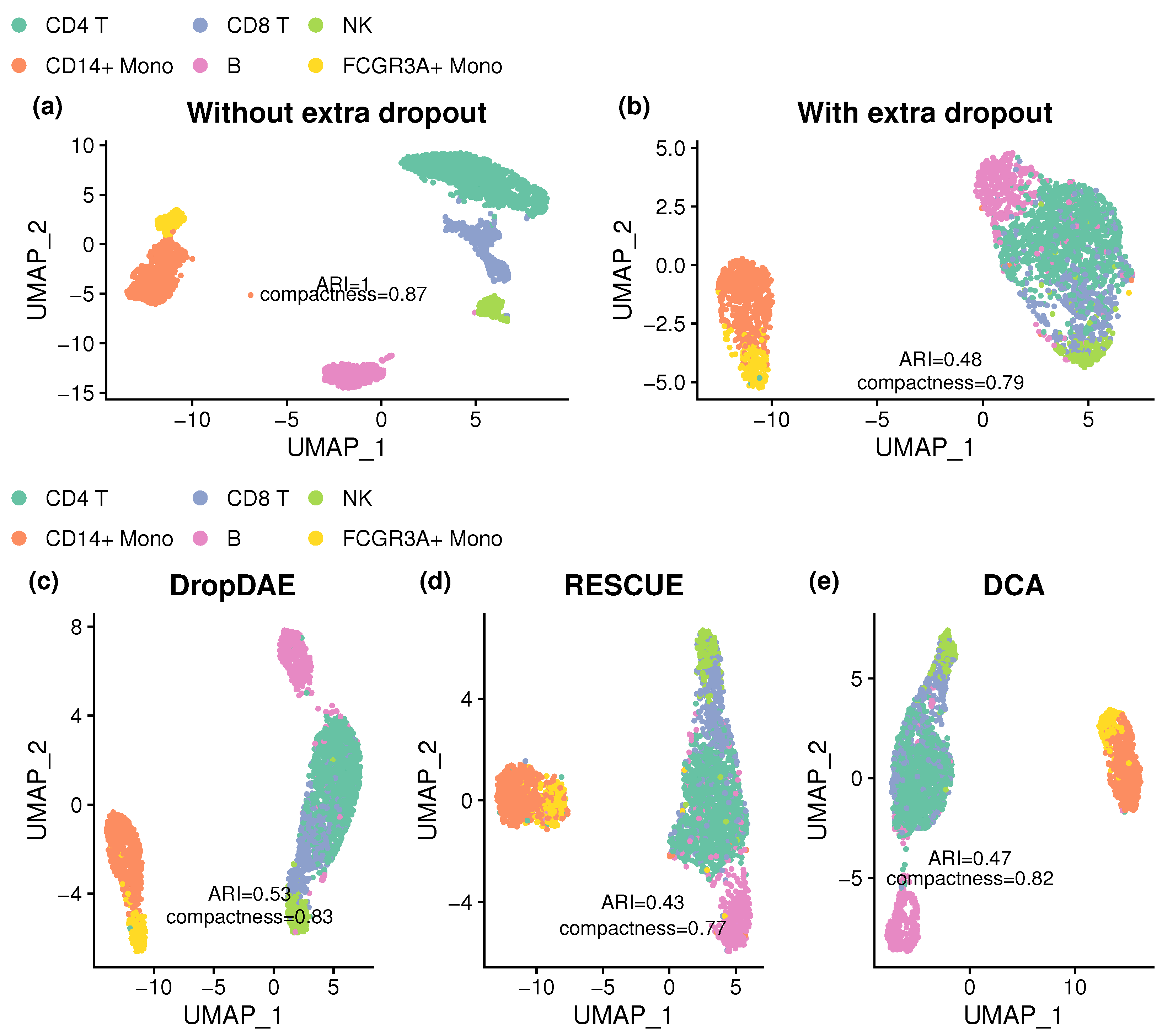

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Interpretation | Value |
|---|---|---|
| seed | Random seed | Index of iteration |
| ngroups | Number of groups | 2 |
| batchCells | Cells per batch | 5000 |
| nGenes | Number of genes | 500 |
| de.prob | DE probability | 0.1 |
| de.facLoc | DE factor location | 0.1 (weak signal) |
| 0.2 (moderate signal) | ||
| 0.3 (strong signal) | ||
| de.facScale | DE factor scale | 0.4 |
| de.downProb | Down-regulation probability | 0.5 |
| dropout.type | Dropout type | “experiment” |
| dropout.mid | Dropout mid point | 2 (weak dropout) |
| 4 (moderate dropout) | ||
| 5 (strong dropout) | ||
| dropout.shape | Dropout shape |
| Method | Two Groups | Six Groups | ||
|---|---|---|---|---|
| ARI | Compactness | ARI | Compactness | |
| Without dropout | 1 (0) | 0.848 (0.015) | 1 (0) | 0.921 (0.004) |
| With dropout | 0.837 (0.197) | 0.551 (0.278) | 0.705 (0.102) | 0.435 (0.304) |
| DropDAE | 0.924 (0.054) | 0.729 (0.065) | 0.81 (0.062) | 0.783 (0.046) |
| DCA | 0.542 (0.264) | 0.45 (0.202) | 0.596 (0.102) | 0.484 (0.256) |
| RESCUE | 0.709 (0.165) | 0.513 (0.21) | 0.483 (0.066) | 0.301 (0.267) |
| Method | ARI | Compactness |
|---|---|---|
| Without dropout | 1 (0) | 0.872 (0) |
| With dropout | 0.475 (0.034) | 0.806 (0.023) |
| DropDAE | 0.558 (0.042) | 0.825 (0.017) |
| RESCUE | 0.414 (0.023) | 0.778 (0.035) |
| DCA | 0.486 (0.034) | 0.803 (0.014) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juan, W.; Ahn, K.W.; Chen, Y.-G.; Lin, C.-W. DropDAE: Denosing Autoencoder with Contrastive Learning for Addressing Dropout Events in scRNA-seq Data. Bioengineering 2025, 12, 829. https://doi.org/10.3390/bioengineering12080829
Juan W, Ahn KW, Chen Y-G, Lin C-W. DropDAE: Denosing Autoencoder with Contrastive Learning for Addressing Dropout Events in scRNA-seq Data. Bioengineering. 2025; 12(8):829. https://doi.org/10.3390/bioengineering12080829
Chicago/Turabian StyleJuan, Wanlin, Kwang Woo Ahn, Yi-Guang Chen, and Chien-Wei Lin. 2025. "DropDAE: Denosing Autoencoder with Contrastive Learning for Addressing Dropout Events in scRNA-seq Data" Bioengineering 12, no. 8: 829. https://doi.org/10.3390/bioengineering12080829
APA StyleJuan, W., Ahn, K. W., Chen, Y.-G., & Lin, C.-W. (2025). DropDAE: Denosing Autoencoder with Contrastive Learning for Addressing Dropout Events in scRNA-seq Data. Bioengineering, 12(8), 829. https://doi.org/10.3390/bioengineering12080829