

Regulation of Tetraspanin CD63 in Chronic Myeloid Leukemia (CML): Single-Cell Analysis of Asymmetric Hematopoietic Stem Cell Division Genes

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Public Datasets
2.1.1. Training Cohort of Transcriptome (GSE4170)
2.1.2. Validation Cohort of RNA Sequencing (GSE100026)
2.1.3. Single-Cell Transcriptome of CML Progenitors (GSE76312)
2.2. Analyses
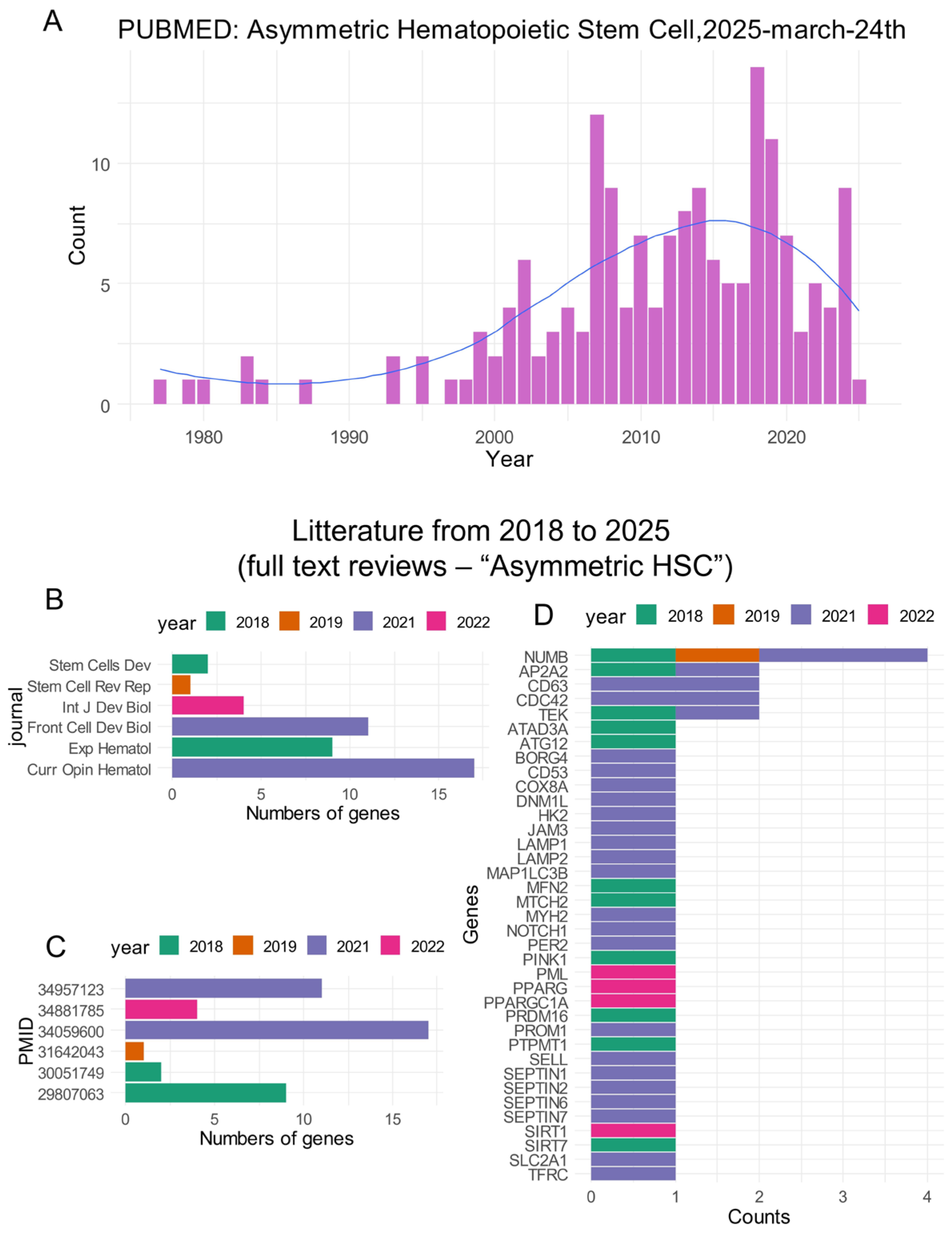
2.2.1. Screening of the Genes in the Literature Related to the Term “Asymmetric Hematopoietic Stem Cell”
2.2.2. Transcriptome Analyses
2.2.3. Single-Cell Asymmetric Hematopoietic Stem Cell Risk Score
2.2.4. Correlation of Single-Cell Artificial Neural Network with Prognosis and Outcome
2.2.5. Single-Cell Trajectory
3. Results
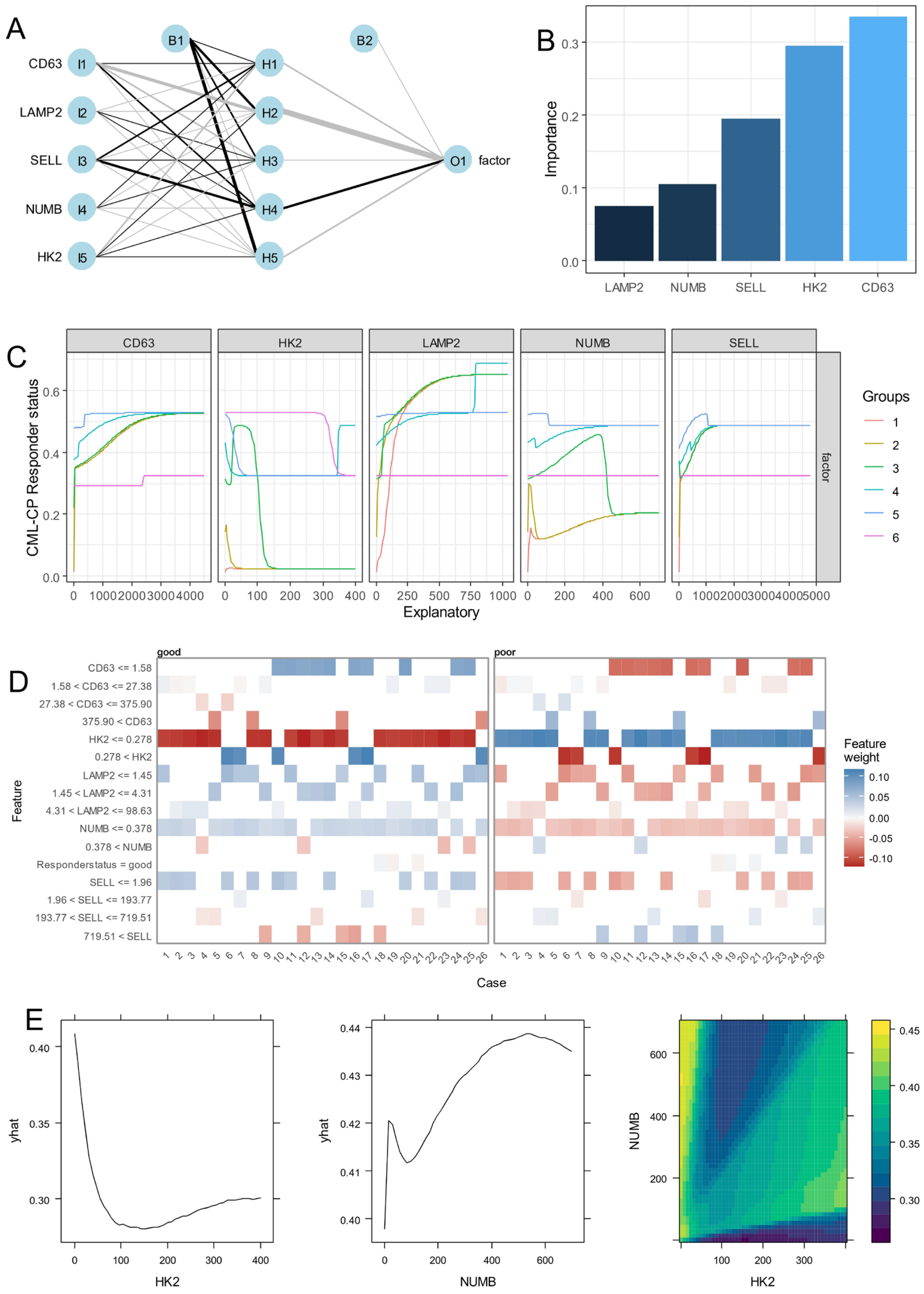
3.1. Identification of Asymmetric Hematopoietic Stem Cell-Related Genes in CML
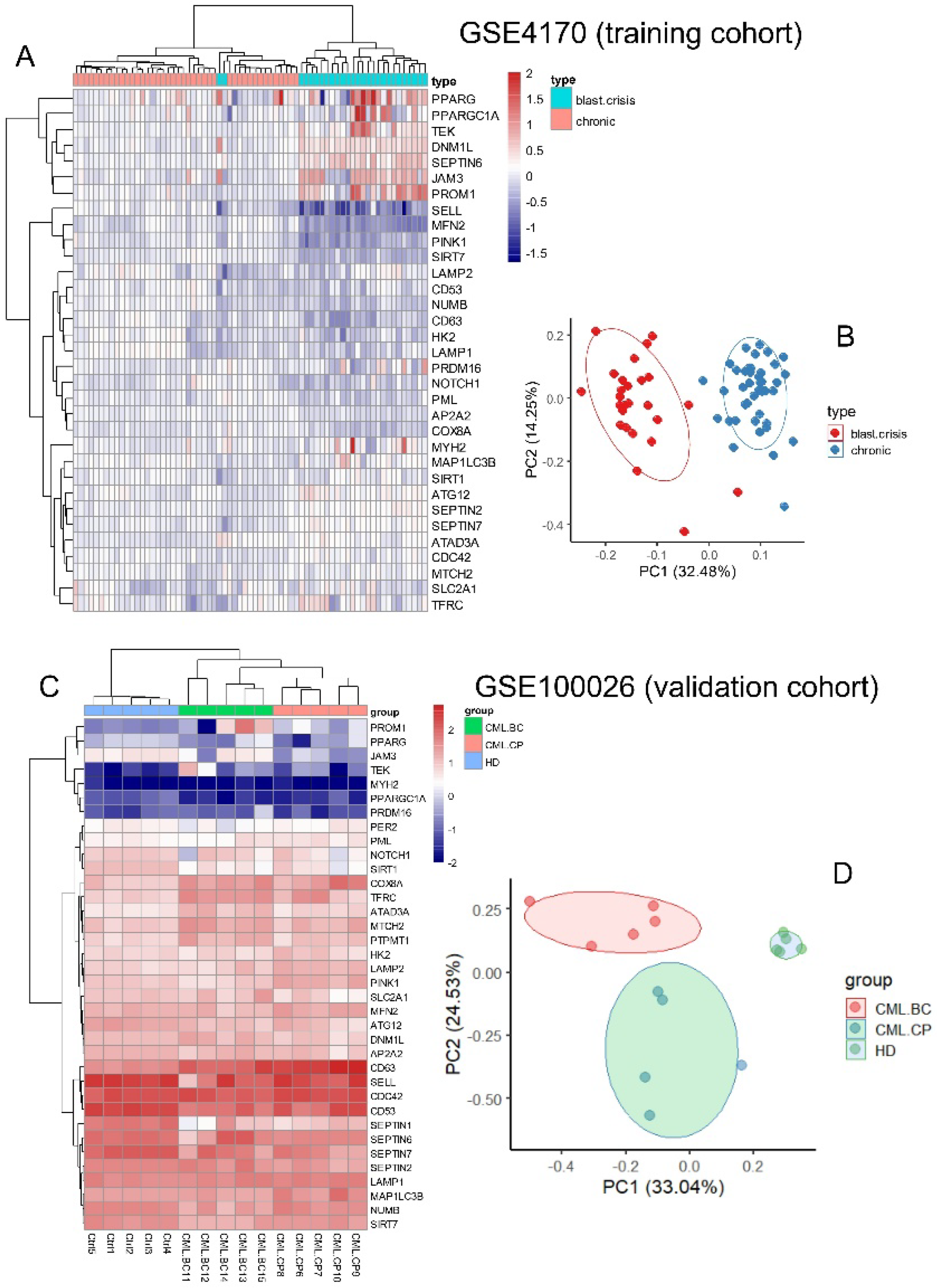
3.2. Expression of Asymmetric Hematopoietic Stem Cell-Related Genes Discriminate Disease Phases During CML Progression
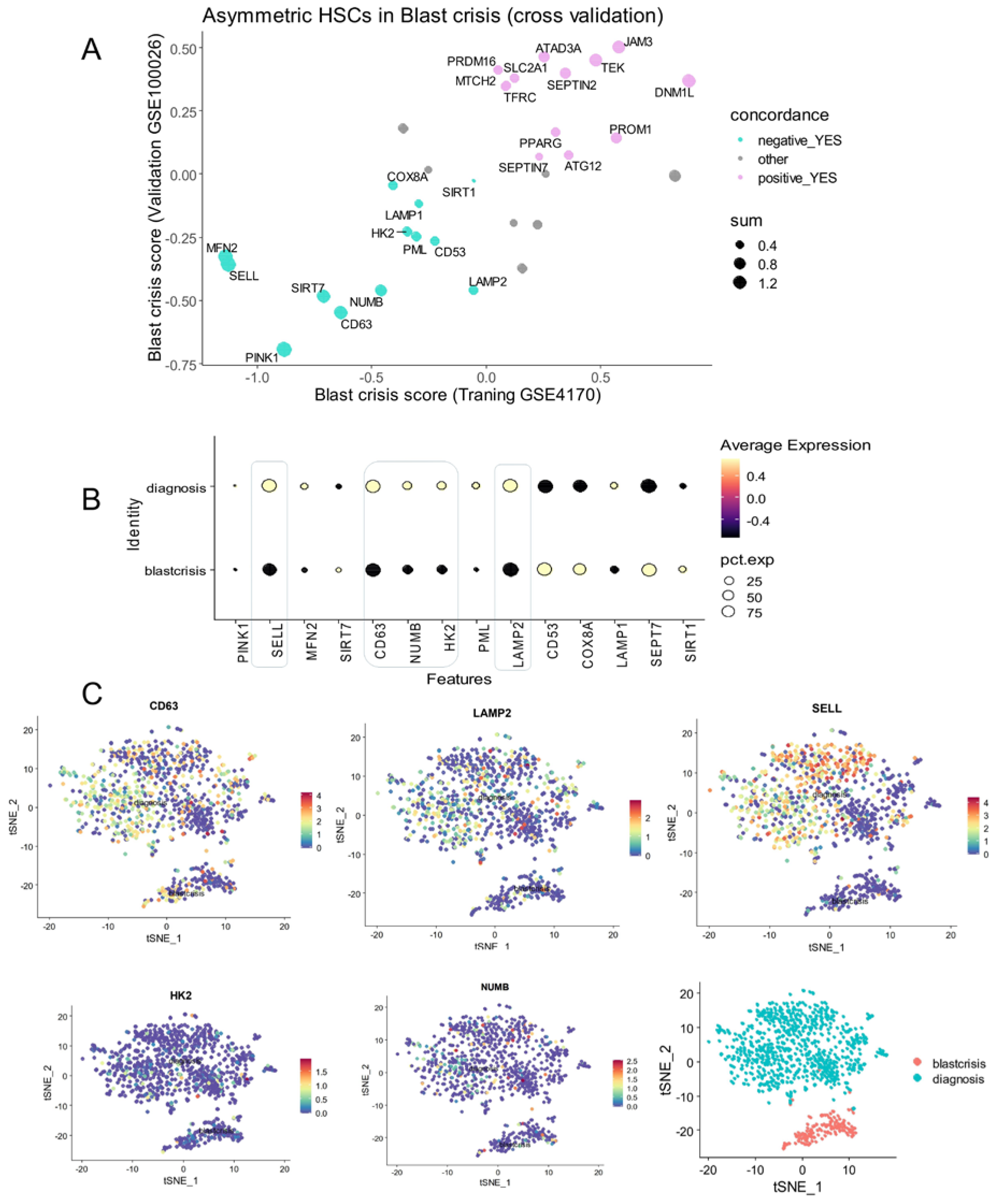
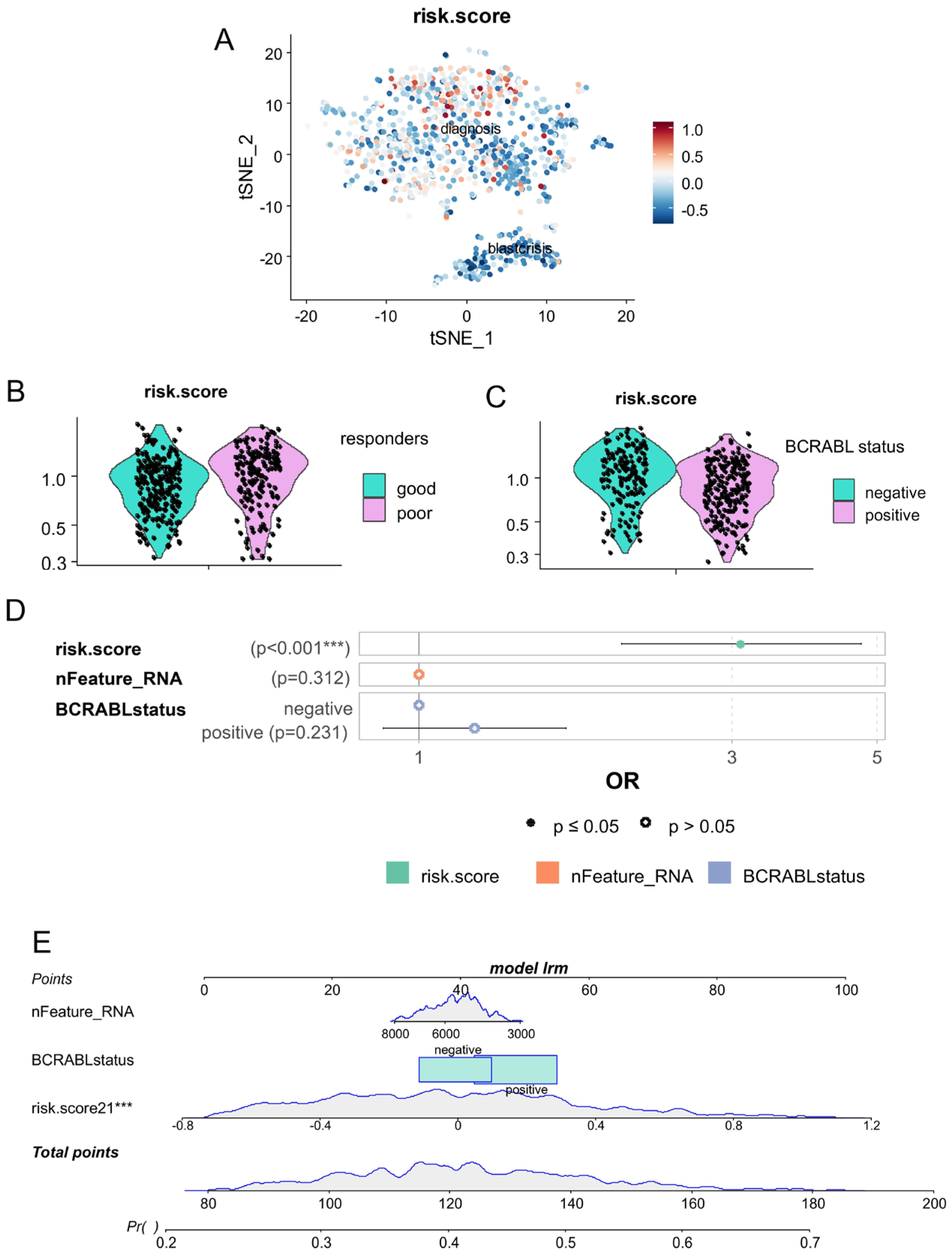
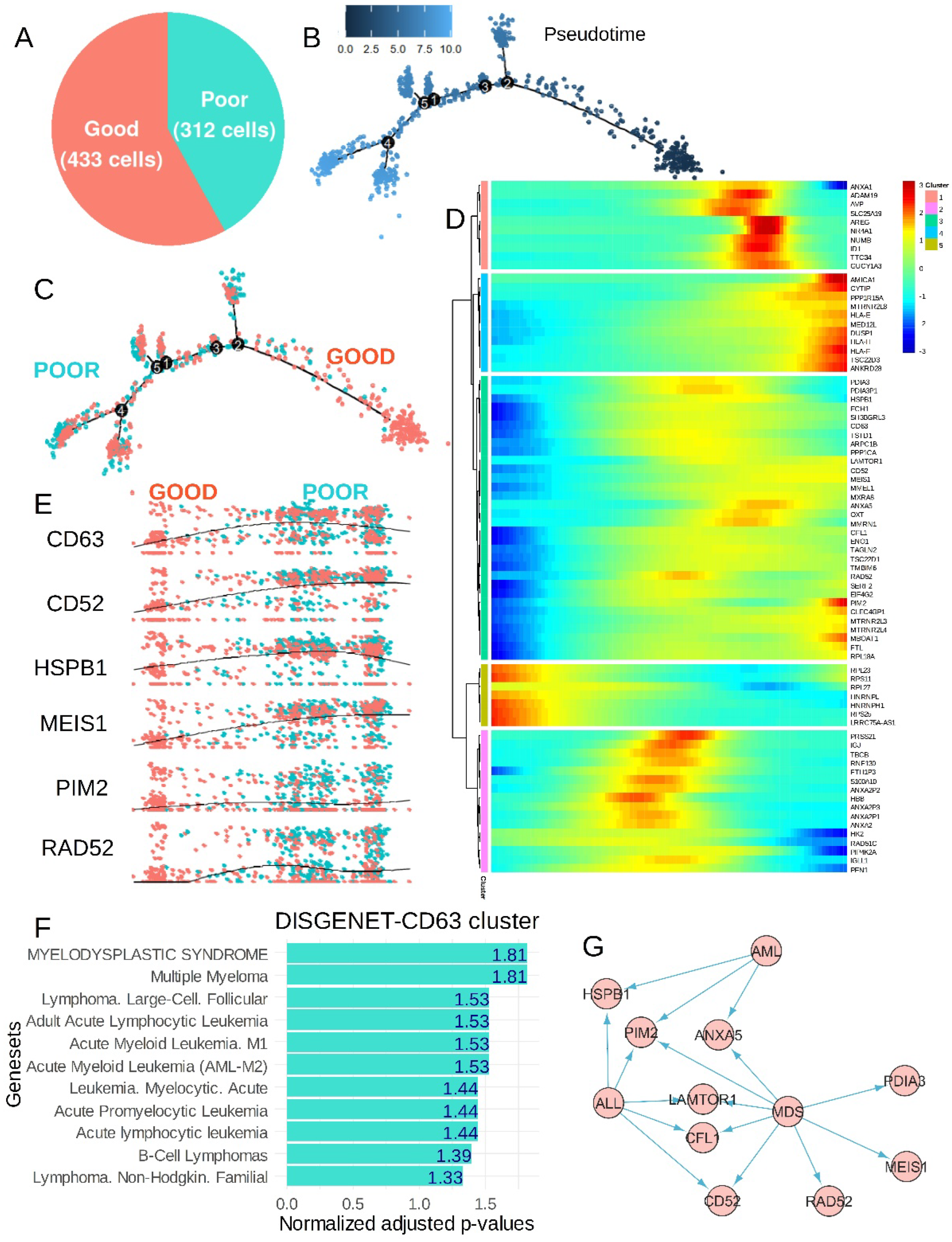
3.3. The CP-CML Single-Cell Trajectory Correlated to the CD63 Expression Profiles of Hematopoietic Malignancies and TKI Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BCR::ABL | Breakpoint cluster region::Abelson murine leukemia viral oncogene homolog 1 |
| CML | Chronic myeloid leukemia |
| CP | Chronic phase |
| BC | Blast crisis |
| ACD | Asymmetric cell divisions |
| MDS | Myelodysplastic syndromes |
| AML | Acute myeloid leukemia |
| ALL | Acute lymphoid leukemia |
| GLUT1 | alias “SLC2A1” for Solute Carrier Family 2 Member 1 |
| TKI | Tyrosine kinase inhibitor |
| PPARG | Peroxisome proliferator activated receptor gamma |
| PPARGC1A | PPARG coactivator 1 alpha |
| TEK | TEK receptor tyrosine kinase |
| DNML1 | Dynamin-1-like protein |
| CD71 | Alias “TFRC”: transferrin receptor |
| CDC42 | Cell division cycle 42 |
| CD34 | Cluster of differentiation 34 |
| CD38 | Cluster of differentiation 38 |
| TP53 | Tumor protein p53 |
| MYC | MYC proto-oncogene, bHLH transcription factor |
| TCF7L2 | Transcription factor 7 like 2 |
| SELL | Selectin L |
| WNT | Wingless-related Integration Site |
| CD63 | Cluster of differentiation 63 |
| NUMB | NUMB endocytic adaptor protein |
| HK2 | Hexokinase 2 |
| LAMP2 | Lsosomal associated membrane protein 2 |
| RAD51C | RAD51 paralog C |
| CD53 | Cluster of differentiation 53 |
| HSPB1 | Heat shock protein family B (small) member 1 |
| HSP27 | Heat Shock Protein 27 |
| PIM2 | Pim-2 proto-oncogene, serine/threonine kinase |
| ANXA5 | Annexin A5 |
| LAMTOR1 | Late endosomal/lysosomal adaptor, MAPK and MTOR activator 1 |
| CFL1 | Cofilin 1 |
| CD52 | Cluster of differentiation 52 |
| RAD52 | RAD52 homolog, DNA repair protein |
| ERCC1 | ERCC excision repair 1, endonuclease non-catalytic subunit |
| MEIS1 | Meis homeobox 1 |
| PDIA3 | Protein disulfide isomerase family A member 3 |
| PBX3 | PBX homeobox 3 |
| ENO1 | Enolase 1 |
| MTRNR2L4 | MT-RNR2 like 4 (pseudogene) |
| MTRNR2L3 | MT-RNR2 like 3 (pseudogene) |
| HIF1α | Hypoxia inducible factor 1 subunit alpha |
| MAPK | Mitogen-activated protein kinases |
| TSC22D1 | TSC22 domain family member 1 |
| TMBIM6 | Transmembrane BAX inhibitor motif containing 6 |
| STAT5 | Signal Transducer and Activator of Transcription 5 |
| STAT4 | Signal Transducer and Activator of Transcription 4 |
| ATAD3A | ATPase family AAA domain containing 3A |
| MFN2 | Mitofusin 2 |
| PINK1 | PTEN induced kinase 1 |
| JAM3 | Junctional adhesion molecule 3 |
| SEPTIN6 | Septin 6 |
| PROM1 | Prominin 1 |
| SIRT7 | Sirtuin 7 |
| PRDM16 | PR/SET domain 16 |
| SEPTIN 2 | Septin 2 |
| AP2A2 | Adaptor related protein complex 2 subunit alpha 2 |
| ID1 | Inhibitor of DNA binding 1, HLH protein |
| NR4A1 | Nuclear receptor subfamily 4 group A member 1 |
| S100A10 | S100 calcium binding protein A10 |
| FTL | Ferritin light chain |
| MLL | Mixed Lineage Leukemia |
| CLL | Chronic lymphocytic leukemia |
| Notch1 | NOTCH receptor 1 |
References
- Goldman, J.M.; Melo, J.V. Chronic Myeloid Leukemia—Advances in Biology and New Approaches to Treatment. N. Engl. J. Med. 2003, 349, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Senapati, J.; Jabbour, E.; Kantarjian, H.; Short, N.J. Pathogenesis and Management of Accelerated and Blast Phases of Chronic Myeloid Leukemia. Leukemia 2023, 37, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene Expression Changes Associated with Progression and Response in Chronic Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Chomel, J.C.; Bonnet, M.-L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.-H.; Ame, S.; et al. Leukemic Stem Cell Persistence in Chronic Myeloid Leukemia Patients in Deep Molecular Response Induced by Tyrosine Kinase Inhibitors and the Impact of Therapy Discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef]
- Chomel, J.-C.; Bonnet, M.-L.; Sorel, N.; Bertrand, A.; Meunier, M.-C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic Stem Cell Persistence in Chronic Myeloid Leukemia Patients with Sustained Undetectable Molecular Residual Disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Müller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic Myeloid Leukemia Stem Cells Are Not Dependent on Bcr-Abl Kinase Activity for Their Survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human Chronic Myeloid Leukemia Stem Cells Are Insensitive to Imatinib despite Inhibition of BCR-ABL Activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef]
- Chomel, J.-C.; Turhan, A.G. Chronic Myeloid Leukemia Stem Cells in the Era of Targeted Therapies: Resistance, Persistence and Long-Term Dormancy. Oncotarget 2011, 2, 713–727. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual Targeting of P53 and C-MYC Selectively Eliminates Leukaemic Stem Cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic Myeloid Leukemia: Mechanisms of Blastic Transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef]
- Desterke, C.; Hugues, P.; Hwang, J.W.; Bennaceur-Griscelli, A.; Turhan, A.G. Embryonic Program Activated during Blast Crisis of Chronic Myelogenous Leukemia (CML) Implicates a TCF7L2 and MYC Cooperative Chromatin Binding. Int. J. Mol. Sci. 2020, 21, 4057. [Google Scholar] [CrossRef]
- Loeffler, D.; Schroeder, T. Symmetric and Asymmetric Activation of Hematopoietic Stem Cells. Curr. Opin. Hematol. 2021, 28, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Scheitza, S.; Wernet, P.; Fischer, J.C.; Giebel, B. Asymmetric Cell Division within the Human Hematopoietic Stem and Progenitor Cell Compartment: Identification of Asymmetrically Segregating Proteins. Blood 2007, 109, 5494–5501. [Google Scholar] [CrossRef]
- Giebel, B.; Zhang, T.; Beckmann, J.; Spanholtz, J.; Wernet, P.; Ho, A.D.; Punzel, M. Primitive Human Hematopoietic Cells Give Rise to Differentially Specified Daughter Cells upon Their Initial Cell Division. Blood 2006, 107, 2146–2152. [Google Scholar] [CrossRef]
- Suda, J.; Suda, T.; Ogawa, M. Analysis of Differentiation of Mouse Hemopoietic Stem Cells in Culture by Sequential Replating of Paired Progenitors. Blood 1984, 64, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Brummendorf, T.H.; Dragowska, W.; Zijlmans, J.M.J.; Thornbury, G.; Lansdorp, P.M. Asymmetric Cell Divisions Sustain Long-Term Hematopoiesis from Single-Sorted Human Fetal Liver Cells. J. Exp. Med. 1998, 188, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Giebel, B.; Beckmann, J. Asymmetric Cell Divisions of Human Hematopoietic Stem and Progenitor Cells Meet Endosomes. Cell Cycle 2007, 6, 2201–2204. [Google Scholar] [CrossRef]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430.e6. [Google Scholar] [CrossRef]
- Loeffler, D.; Schneiter, F.; Wang, W.; Wehling, A.; Kull, T.; Lengerke, C.; Manz, M.G.; Schroeder, T. Asymmetric Organelle Inheritance Predicts Human Blood Stem Cell Fate. Blood 2022, 139, 2011–2023. [Google Scholar] [CrossRef]
- Wilson, A.; Ardiet, D.-L.; Saner, C.; Vilain, N.; Beermann, F.; Aguet, M.; Macdonald, H.R.; Zilian, O. Normal Hemopoiesis and Lymphopoiesis in the Combined Absence of Numb and Numblike. J. Immunol. 2007, 178, 6746–6751. [Google Scholar] [CrossRef]
- Florian, M.C.; Klose, M.; Sacma, M.; Jablanovic, J.; Knudson, L.; Nattamai, K.J.; Marka, G.; Vollmer, A.; Soller, K.; Sakk, V.; et al. Aging Alters the Epigenetic Asymmetry of HSC Division. PLoS Biol. 2018, 16, e2003389. [Google Scholar] [CrossRef]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.-D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 Activity Regulates Hematopoietic Stem Cell Aging and Rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kwon, H.Y.; Zimdahl, B.; Congdon, K.L.; Blum, J.; Lento, W.E.; Zhao, C.; Lagoo, A.; Gerrard, G.; Foroni, L.; et al. Regulation of Myeloid Leukaemia by the Cell-Fate Determinant Musashi. Nature 2010, 466, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Kharas, M.G.; Lengner, C.J.; Al-Shahrour, F.; Bullinger, L.; Ball, B.; Zaidi, S.; Morgan, K.; Tam, W.; Paktinat, M.; Okabe, R.; et al. Musashi-2 Regulates Normal Hematopoiesis and Promotes Aggressive Myeloid Leukemia. Nat. Med. 2010, 16, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Mizukawa, B.; O’Brien, E.; Moreira, D.C.; Wunderlich, M.; Hochstetler, C.L.; Duan, X.; Liu, W.; Orr, E.; Grimes, H.L.; Mulloy, J.C.; et al. The Cell Polarity Determinant CDC42 Controls Division Symmetry to Block Leukemia Cell Differentiation. Blood 2017, 130, 1336–1346. [Google Scholar] [CrossRef]
- Girotra, M.; Trachsel, V.; Roch, A.; Lutolf, M.P. In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro. Int. J. Mol. Sci. 2020, 21, 8225. [Google Scholar] [CrossRef]
- Thomas, G.E.; Egan, G.; García-Prat, L.; Botham, A.; Voisin, V.; Patel, P.S.; Hoff, F.W.; Chin, J.; Nachmias, B.; Kaufmann, K.B.; et al. The Metabolic Enzyme Hexokinase 2 Localizes to the Nucleus in AML and Normal Haematopoietic Stem and Progenitor Cells to Maintain Stemness. Nat. Cell Biol. 2022, 24, 872–884. [Google Scholar] [CrossRef]
- Zeng, Z.; Sarbassov, D.D.; Samudio, I.J.; Yee, K.W.L.; Munsell, M.F.; Ellen Jackson, C.; Giles, F.J.; Sabatini, D.M.; Andreeff, M.; Konopleva, M. Rapamycin Derivatives Reduce mTORC2 Signaling and Inhibit AKT Activation in AML. Blood 2007, 109, 3509–3512. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Li, S.-Q.; Liu, J.; Zhang, J.; Wang, X.-L.; Chen, D.; Wang, Y.; Xu, Y.-M.; Huang, B.; Lin, J.; Li, J.; et al. Transcriptome Profiling Reveals the High Incidence of hnRNPA1 Exon 8 Inclusion in Chronic Myeloid Leukemia. J. Adv. Res. 2020, 24, 301–310. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering Splice Junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-Cell Transcriptomics Uncovers Distinct Molecular Signatures of Stem Cells in Chronic Myeloid Leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37, D5–D15. [Google Scholar] [CrossRef]
- Wei, C.-H.; Allot, A.; Leaman, R.; Lu, Z. PubTator Central: Automated Concept Annotation for Biomedical Full Text Articles. Nucleic Acids Res. 2019, 47, W587–W593. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of Multiple Cancer Types by Shrunken Centroids of Gene Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef]
- Beck, M.W. NeuralNetTools: Visualization and Analysis Tools for Neural Networks. J. Stat. Softw. 2018, 85, 1–20. [Google Scholar] [CrossRef]
- Zhang, Z.; Beck, M.W.; Winkler, D.A.; Huang, B.; Sibanda, W.; Goyal, H.; written on behalf of AME Big-Data Clinical Trial Collaborative Group. Opening the Black Box of Neural Networks: Methods for Interpreting Neural Network Models in Clinical Applications. Ann. Transl. Med. 2018, 6, 216. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Qiu, X.; Trapnell, C.; Mao, Q.; Wang, L. DDRTree: Learning Principal Graphs with DDRTree. Rcpp 2017. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of Biological Networks and Gene Expression Data Using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef]
- Shen, Q.; Temple, S. Creating Asymmetric Cell Divisions by Skewing Endocytosis. Sci. STKE Signal Transduct. Knowl. Environ. 2002, 2002, pe52. [Google Scholar] [CrossRef]
- Borsa, M.; Barnstorf, I.; Baumann, N.S.; Pallmer, K.; Yermanos, A.; Gräbnitz, F.; Barandun, N.; Hausmann, A.; Sandu, I.; Barral, Y.; et al. Modulation of Asymmetric Cell Division as a Mechanism to Boost CD8+ T Cell Memory. Sci. Immunol. 2019, 4, eaav1730. [Google Scholar] [CrossRef]
- Eisterer, W.; Jiang, X.; Christ, O.; Glimm, H.; Lee, K.H.; Pang, E.; Lambie, K.; Shaw, G.; Holyoake, T.L.; Petzer, A.L.; et al. Different Subsets of Primary Chronic Myeloid Leukemia Stem Cells Engraft Immunodeficient Mice and Produce a Model of the Human Disease. Leukemia 2005, 19, 435–441. [Google Scholar] [CrossRef]
- Jiang, X.; Forrest, D.; Nicolini, F.; Turhan, A.; Guilhot, J.; Yip, C.; Holyoake, T.; Jorgensen, H.; Lambie, K.; Saw, K.M.; et al. Properties of CD34+ CML Stem/Progenitor Cells That Correlate with Different Clinical Responses to Imatinib Mesylate. Blood 2010, 116, 2112–2121. [Google Scholar] [CrossRef]
- Pols, M.S.; Klumperman, J. Trafficking and Function of the Tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Hu, M.; Lu, Y.; Wang, S.; Zhang, Z.; Qi, Y.; Chen, N.; Shen, M.; Chen, F.; Chen, M.; Yang, L.; et al. CD63 Acts as a Functional Marker in Maintaining Hematopoietic Stem Cell Quiescence through Supporting TGFβ Signaling in Mice. Cell Death Differ. 2022, 29, 178–191. [Google Scholar] [CrossRef]
- Berdnik, D.; Török, T.; González-Gaitán, M.; Knoblich, J.A. The Endocytic Protein Alpha-Adaptin Is Required for Numb-Mediated Asymmetric Cell Division in Drosophila. Dev. Cell 2002, 3, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T Cell Fate Specification in Mice with an Induced Inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Hattori, M.; Nakamura, E.; Nakanishi, S.; Minato, N.; Kageyama, R. The bHLH Gene Hes1 Is Essential for Expansion of Early T Cell Precursors. Genes Dev. 1999, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Pui, J.C.; Allman, D.; Xu, L.; DeRocco, S.; Karnell, F.G.; Bakkour, S.; Lee, J.Y.; Kadesch, T.; Hardy, R.R.; Aster, J.C.; et al. Notch1 Expression in Early Lymphopoiesis Influences B versus T Lineage Determination. Immunity 1999, 11, 299–308. [Google Scholar] [CrossRef]
- Wilson, A.; MacDonald, H.R.; Radtke, F. Notch 1-Deficient Common Lymphoid Precursors Adopt a B Cell Fate in the Thymus. J. Exp. Med. 2001, 194, 1003–1012. [Google Scholar] [CrossRef]
- French, M.B.; Koch, U.; Shaye, R.E.; McGill, M.A.; Dho, S.E.; Guidos, C.J.; McGlade, C.J. Transgenic Expression of Numb Inhibits Notch Signaling in Immature Thymocytes but Does Not Alter T Cell Fate Specification. J. Immunol. 2002, 168, 3173–3180. [Google Scholar] [CrossRef]
- Yang, L.; Cao, L.; Yang, M.; Tang, D.; Kang, R.; Min, X.; Zhu, S.; Yu, Y. Hsp27: A Novel Therapeutic Target for Pediatric M4/M5 Acute Myeloid Leukemia. Oncol. Rep. 2013, 29, 1459–1466. [Google Scholar] [CrossRef]
- Ma, L.; Pak, M.L.; Ou, J.; Yu, J.; St Louis, P.; Shan, Y.; Hutchinson, L.; Li, S.; Brehm, M.A.; Zhu, L.J.; et al. Prosurvival Kinase PIM2 Is a Therapeutic Target for Eradication of Chronic Myeloid Leukemia Stem Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10482–10487. [Google Scholar] [CrossRef]
- Yang, J.; Liu, P.; Ma, D.; Zhao, P.; Zhang, Y.; Lu, Y.; Li, Y.; Huang, Y.; Chen, Y.; Wang, J. Glucocorticoid Resistance Induced by ANXA5 Overexpression in B-Cell Acute Lymphoblastic Leukemia. Pediatr. Hematol. Oncol. 2021, 38, 36–48. [Google Scholar] [CrossRef]
- Yonehara, R.; Nada, S.; Nakai, T.; Nakai, M.; Kitamura, A.; Ogawa, A.; Nakatsumi, H.; Nakayama, K.I.; Li, S.; Standley, D.M.; et al. Structural Basis for the Assembly of the Ragulator-Rag GTPase Complex. Nat. Commun. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Valiulienė, G.; Vitkevičienė, A.; Navakauskienė, R. The Epigenetic Treatment Remodel Genome-Wide Histone H4 Hyper-Acetylation Patterns and Affect Signaling Pathways in Acute Promyelocytic Leukemia Cells. Eur. J. Pharmacol. 2020, 889, 173641. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Ji, X.; Lei, Y.; Jia, Y.; Liu, F.; Xia, H.; Tan, H.; Zeng, X.; Yi, L.; He, J.; et al. Diallyl Disulfide Induces Downregulation and Inactivation of Cofilin 1 Differentiation via the Rac1/ROCK1/LIMK1 Pathway in Leukemia Cells. Int. J. Oncol. 2020, 56, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Li, L.; Haak, M.; Brors, B.; Frank, O.; Giehl, M.; Fabarius, A.; Schatz, M.; Weisser, A.; Lorentz, C.; et al. Gene Expression Profiling of CD34+ Cells Identifies a Molecular Signature of Chronic Myeloid Leukemia Blast Crisis. Leukemia 2006, 20, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Loeff, F.C.; Falkenburg, J.H.F.; Hageman, L.; Huisman, W.; Veld, S.A.J.; van Egmond, H.M.E.; van de Meent, M.; von dem Borne, P.A.; Veelken, H.; Halkes, C.J.M.; et al. High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI Anchor-/CD52- T Cells That Can Give Early Immune Protection after Alemtuzumab-Based T Cell-Depleted Allogeneic Stem Cell Transplantation. J. Immunol. 2018, 200, 2199–2208. [Google Scholar] [CrossRef]
- Dias, A.; Kenderian, S.J.; Westin, G.F.; Litzow, M.R. Novel Therapeutic Strategies in Acute Lymphoblastic Leukemia. Curr. Hematol. Malign Rep. 2016, 11, 253–264. [Google Scholar] [CrossRef]
- Rodig, S.J.; Abramson, J.S.; Pinkus, G.S.; Treon, S.P.; Dorfman, D.M.; Dong, H.Y.; Shipp, M.A.; Kutok, J.L. Heterogeneous CD52 Expression among Hematologic Neoplasms: Implications for the Use of Alemtuzumab (CAMPATH-1H). Clin. Cancer Res. 2006, 12, 7174–7179. [Google Scholar] [CrossRef]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.P.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and Other Kinases from Chronic Myeloproliferative Disorders Stimulate Single-Strand Annealing, an Unfaithful DNA Double-Strand Break Repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 Regulates the Metabolic Phenotype and Oxidant Defense of Hematopoietic Stem Cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef]
- Li, Z.; Chen, P.; Su, R.; Hu, C.; Li, Y.; Elkahloun, A.G.; Zuo, Z.; Gurbuxani, S.; Arnovitz, S.; Weng, H.; et al. PBX3 and MEIS1 Cooperate in Hematopoietic Cells to Drive Acute Myeloid Leukemias Characterized by a Core Transcriptome of the MLL-Rearranged Disease. Cancer Res. 2016, 76, 619–629. [Google Scholar] [CrossRef]
- Ye, Q.; Fu, P.; Dou, J.; Wang, N. Downregulation of PDIA3 Inhibits Proliferation and Invasion of Human Acute Myeloid Leukemia Cells. OncoTargets Ther. 2018, 11, 2925–2935. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desterke, C.; Bennaceur-Griscelli, A.; Turhan, A.G. Regulation of Tetraspanin CD63 in Chronic Myeloid Leukemia (CML): Single-Cell Analysis of Asymmetric Hematopoietic Stem Cell Division Genes. Bioengineering 2025, 12, 830. https://doi.org/10.3390/bioengineering12080830
Desterke C, Bennaceur-Griscelli A, Turhan AG. Regulation of Tetraspanin CD63 in Chronic Myeloid Leukemia (CML): Single-Cell Analysis of Asymmetric Hematopoietic Stem Cell Division Genes. Bioengineering. 2025; 12(8):830. https://doi.org/10.3390/bioengineering12080830
Chicago/Turabian StyleDesterke, Christophe, Annelise Bennaceur-Griscelli, and Ali G. Turhan. 2025. "Regulation of Tetraspanin CD63 in Chronic Myeloid Leukemia (CML): Single-Cell Analysis of Asymmetric Hematopoietic Stem Cell Division Genes" Bioengineering 12, no. 8: 830. https://doi.org/10.3390/bioengineering12080830
APA StyleDesterke, C., Bennaceur-Griscelli, A., & Turhan, A. G. (2025). Regulation of Tetraspanin CD63 in Chronic Myeloid Leukemia (CML): Single-Cell Analysis of Asymmetric Hematopoietic Stem Cell Division Genes. Bioengineering, 12(8), 830. https://doi.org/10.3390/bioengineering12080830