

An Integrated In Vivo/In Vitro Protein Production Platform for Site-Specific Antibody Drug Conjugates

 ,
,
Abstract
1. Introduction
2. Materials and Methods
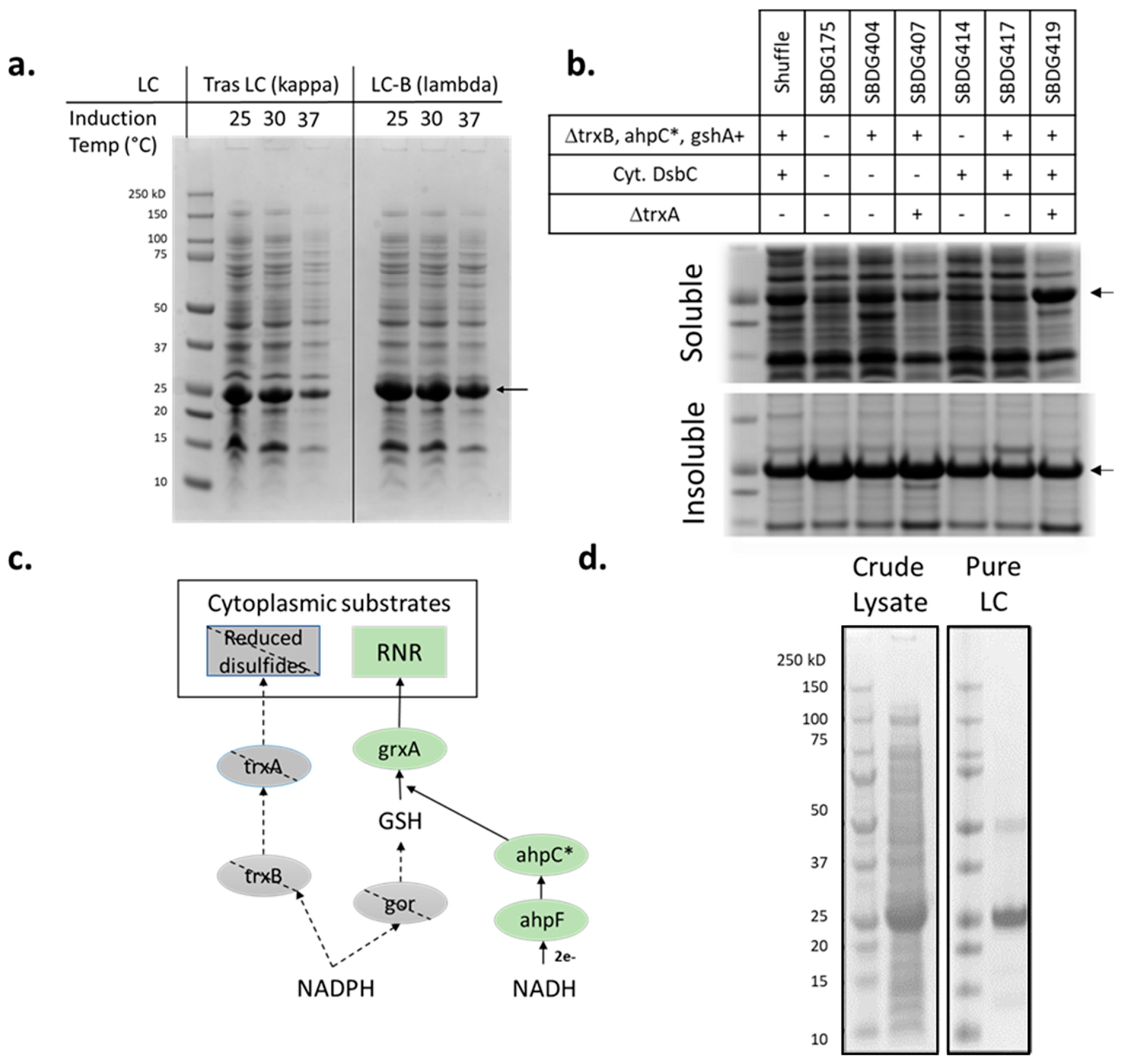
2.1. Light-Chain Construct Design
2.2. Strain Engineering
2.3. Light-Chain Shake Flask Expression
2.4. Light-Chain Bioreactor Expression
2.5. Light-Chain Purification
2.6. mAb Expression in Cell-Free Reactions
2.7. mAb Purification and Conjugation
2.8. In Vitro Cell Killing Assays
2.9. ADC Binding to FcRn by Bio-Layer Interferometry
2.10. LC-MS Analysis of ADCs
3. Results and Discussion
3.1. Light-Chain Expression in E. coli

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Citation |
|---|---|---|
| Shuffle® T7 | F’ lac, pro, lacIq/Δ(ara-leu)7697 araD139 fhuA2 lacZ::T7 gene1 Δ(phoA)PvuII phoR ahpC* galE (or U) galK λatt::pNEB3-r1-cDsbC (SpecR, lacIq) ΔtrxB rpsL150(StrR) Δgor Δ(malF)3 | [25] |
| SBHS016 | A19 ΔtonA ΔtnaA ΔspeA ΔendA ΔLAM ΔsdaA ΔsdaB ΔgshA Δgor RF1(N296K/L297R/L298R) araB::T7 galK::Pc0-2XDsbC met+ | [6] |
| SBDG175 | SBHS016 ΔompT araB::T7 galK::Pc0-2XDsbC | this work |
| SBDG404 | SBSH016 araB::T7 galK::Pc0-2XDsbC tnaA::Pc0-ahpC* trxB::Pwt-gshA | this work |
| SBDG407 | SBDG403 ΔtrxA | this work |
| SBDG414 | SBHS016 lacZ::Pmtl-2xlDsbC | this work |
| SBDG417 | SBDG404 lacZ::Pmtl-2xlDsbC | this work |
| SBDG419 | SBDG407 lacZ::Pmtl-2xlDsbC | this work |
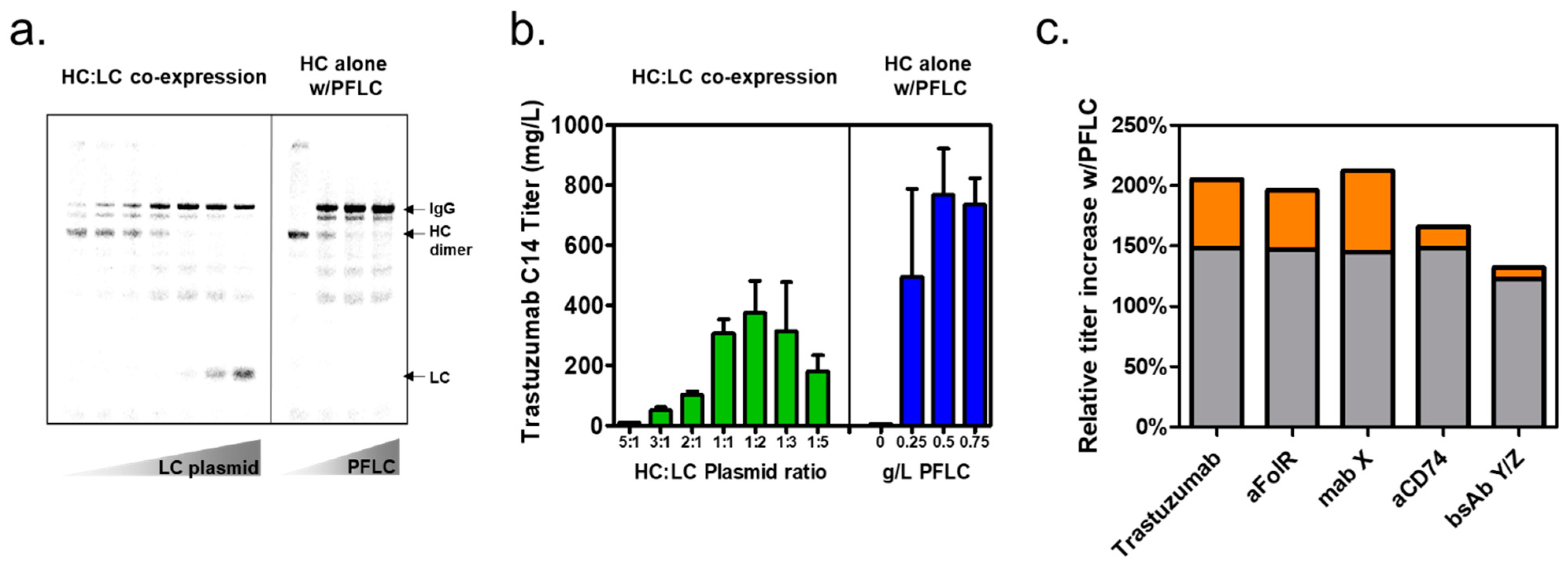
3.2. Expression of Trastuzumab IgG with Prefabricated Light Chain in XpressCF+®
3.3. Prefabricated Light-Chain Titer Improvements for Additional Antibodies
3.4. Demonstration of Prefabricated Light-Chain Titer Improvements at Larger Reaction Formats
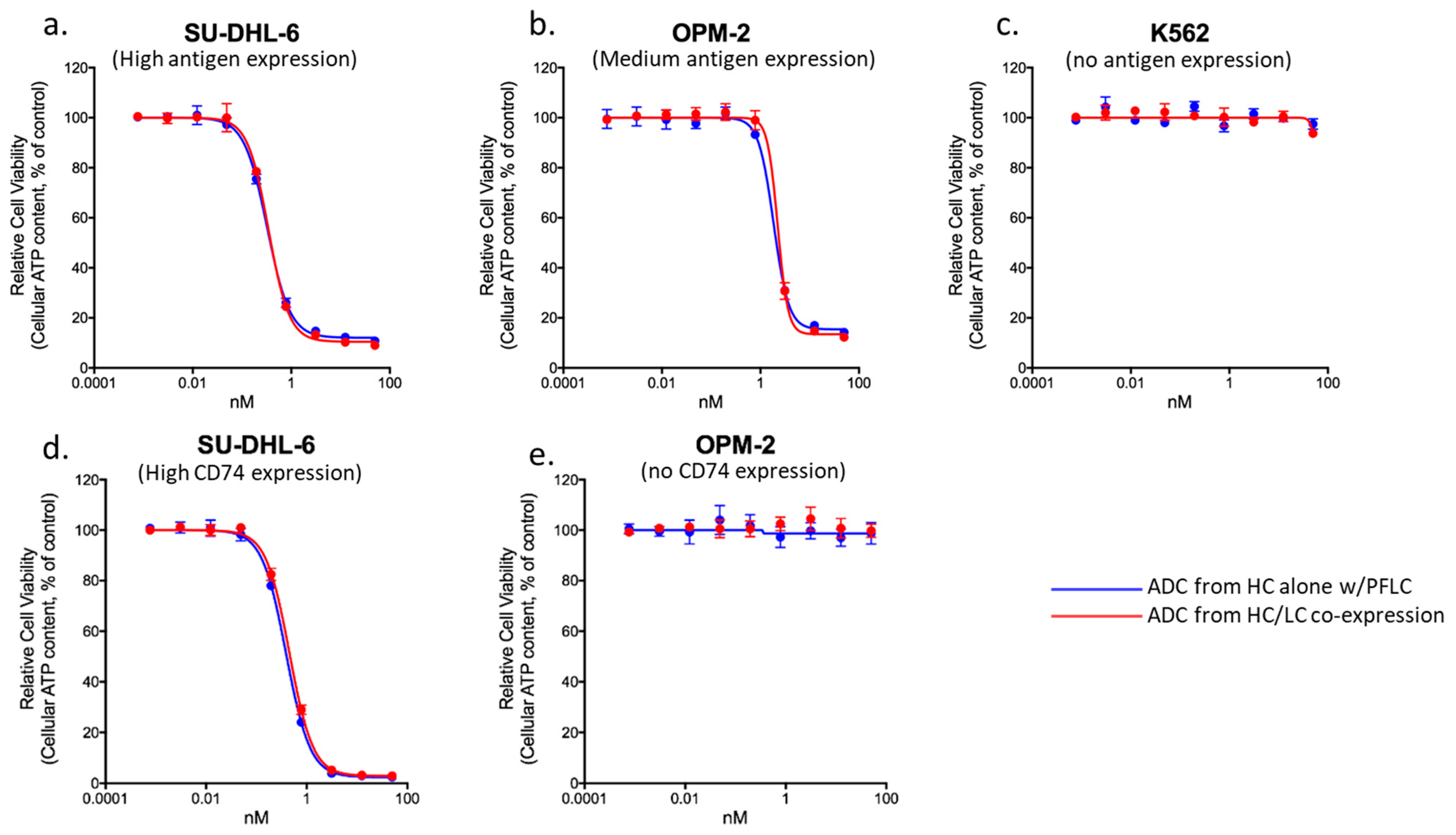
3.5. Comparability of ADCs Produced with Prefabricated Light Chain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Kline, T.; Steiner, A.R.; Penta, K.; Sato, A.K.; Hallam, T.J.; Yin, G. Methods to Make Homogenous Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3480–3493. [Google Scholar] [CrossRef]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of Site-Specific Antibody–Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef]
- Yin, G.; Stephenson, H.T.; Yang, J.; Li, X.; Armstrong, S.M.; Heibeck, T.H.; Tran, C.; Masikat, M.R.; Zhou, S.; Stafford, R.L.; et al. RF1 attenuation enables efficient non-natural amino acid incorporation for production of homogeneous antibody drug conjugates. Sci. Rep. 2017, 7, 3026. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef]
- Zawada, J.F.; Burgenson, D.; Yin, G.; Hallam, T.J.; Swartz, J.R.; Kiss, R.D. Cell-free technologies for biopharmaceutical research and production. Curr. Opin. Biotechnol. 2022, 76, 102719. [Google Scholar] [CrossRef]
- Schmied, W.H.; Elsässer, S.J.; Uttamapinant, C.; Chin, J.W. Efficient Multisite Unnatural Amino Acid Incorporation in Mammalian Cells via Optimized Pyrrolysyl tRNA Synthetase/tRNA Expression and Engineered eRF1. J. Am. Chem. Soc. 2014, 136, 15577–15583. [Google Scholar] [CrossRef]
- Roy, G.; Reier, J.; Garcia, A.; Martin, T.; Rice, M.; Wang, J.; Prophet, M.; Christie, R.; Dall’Acqua, W.; Ahuja, S.; et al. Development of a high yielding expression platform for the introduction of non-natural amino acids in protein sequences. Mabs 2020, 12, 1684749. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.F.; Xu, J.; Shen, Z.; Takimoto, J.K.; Schultz, M.D.; Schmitz, R.; Xiang, Z.; Ecker, J.; Briggs, S.P.; Wang, L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically Recoded Organisms Expand Biological Functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef]
- Robinson, M.-P.; Ke, N.; Lobstein, J.; Peterson, C.; Szkodny, A.; Mansell, T.J.; Tuckey, C.; Riggs, P.D.; Colussi, P.A.; Noren, C.J.; et al. Efficient expression of full-length antibodies in the cytoplasm of engineered bacteria. Nat. Commun. 2015, 6, 8072. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Garces, E.D.; Yang, J.; Zhang, J.; Tran, C.; Steiner, A.R.; Roos, C.; Bajad, S.; Hudak, S.; Penta, K.; et al. Aglycosylated antibodies and antibody fragments produced in a scalable in vitro transcription-translation system. mAbs 2012, 4, 217–225. [Google Scholar] [CrossRef]
- Yin, G.; Swartz, J.R. Enhancing multiple disulfide bonded protein folding in a cell-free system. Biotechnol. Bioeng. 2004, 86, 188–195. [Google Scholar] [CrossRef]
- Groff, D.; Armstrong, S.; Rivers, P.J.; Zhang, J.; Yang, J.; Green, E.; Rozzelle, J.; Liang, S.; Kittle, J.D.; Steiner, A.R.; et al. Engineering toward a bacterial “endoplasmic reticulum” for the rapid expression of immunoglobulin proteins. mAbs 2014, 6, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Michel-Reydellet, N.; Calhoun, K.; Swartz, J. Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metab. Eng. 2004, 6, 197–203. [Google Scholar] [CrossRef]
- Cole, S.D.; Miklos, A.E.; Chiao, A.C.; Sun, Z.Z.; Lux, M.W. Methodologies for preparation of prokaryotic extracts for cell-free expression systems. Synth. Syst. Biotechnol. 2020, 5, 252–267. [Google Scholar] [CrossRef]
- Dopp, B.J.L.; Tamiev, D.D.; Reuel, N.F. Cell-free supplement mixtures: Elucidating the history and biochemical utility of additives used to support in vitro protein synthesis in E. coli extract. Biotechnol. Adv. 2019, 37, 246–258. [Google Scholar] [CrossRef]
- Cai, Q.; Hanson, J.A.; Steiner, A.R.; Tran, C.; Masikat, M.R.; Chen, R.; Zawada, J.F.; Sato, A.K.; Hallam, T.J.; Yin, G. A simplified and robust protocol for immunoglobulin expression in Escherichia coli cell-free protein synthesis systems. Biotechnol. Prog. 2015, 31, 823–831. [Google Scholar] [CrossRef]
- Borkowski, O.; Koch, M.; Zettor, A.; Pandi, A.; Batista, A.C.; Soudier, P.; Faulon, J.-L. Large scale active-learning-guided exploration for in vitro protein production optimization. Nat. Commun. 2020, 11, 1872. [Google Scholar] [CrossRef] [PubMed]
- Stafford, R.L.; Matsumoto, M.; Yin, G.; Cai, Q.; Fung, J.J.; Stephenson, H.; Gill, A.; You, M.; Lin, S.-H.; Wang, W.D.; et al. In vitro Fab display: A cell-free system for IgG discovery. Protein Eng. Des. Sel. 2014, 27, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Zawada, J.F.; Yin, G.; Steiner, A.R.; Yang, J.; Naresh, A.; Roy, S.M.; Gold, D.S.; Heinsohn, H.G.; Murray, C.J. Microscale to manufacturing scale-up of cell-free cytokine production—A new approach for shortening protein production development timelines. Biotechnol. Bioeng. 2011, 108, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Factories 2012, 11, 753. [Google Scholar] [CrossRef]
- Bessette, P.H.; Åslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef]
- Ortenberg, R.; Gon, S.; Porat, A.; Beckwith, J. Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 7439–7444. [Google Scholar] [CrossRef]
- Ritz, D.; Lim, J.; Reynolds, C.M.; Poole, L.B.; Beckwith, J. Conversion of a Peroxiredoxin into a Disulfide Reductase by a Triplet Repeat Expansion. Science 2001, 294, 158–160. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ritz, D.; Planson, A.-G.; Jönsson, T.J.; Faulkner, M.J.; Boyd, D.; Beckwith, J.; Poole, L.B. Mutant AhpC peroxiredoxins suppress thiol-disulfide redox deficiencies and acquire deglutathionylating activity. Mol. Cell 2008, 29, 36–45. [Google Scholar] [CrossRef]
- Calhoun, K.A.; Swartz, J.R. Total amino acid stabilization during cell-free protein synthesis reactions. J. Biotechnol. 2006, 123, 193–203. [Google Scholar] [CrossRef]
- Knapp, K.G.; Swartz, J.R. Evidence for an additional disulfide reduction pathway in Escherichia coli. J. Biosci. Bioeng. 2007, 103, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Groff, D.; Carlos, N.A.; Chen, R.; Hanson, J.A.; Liang, S.; Armstrong, S.; Li, X.; Zhou, S.; Steiner, A.; Hallam, T.J.; et al. Development of an E. coli strain for cell-free ADC manufacturing. Biotechnol. Bioeng. 2022, 119, 162–175. [Google Scholar] [CrossRef]
- Feige, M.J.; Groscurth, S.; Marcinowski, M.; Shimizu, Y.; Kessler, H.; Hendershot, L.M.; Buchner, J. An Unfolded CH1 Domain Controls the Assembly and Secretion of IgG Antibodies. Mol. Cell 2009, 34, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Abrahams, C.L.; Li, X.; Embry, M.; Yu, A.; Krimm, S.; Krueger, S.; Greenland, N.Y.; Wen, K.W.; Jones, C.; DeAlmeida, V.; et al. Targeting CD74 in multiple myeloma with the novel, site-specific antibody-drug conjugate STRO-001. Oncotarget 2018, 9, 37700–37714. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Le, T.; Rock, B.M.; Rock, D.A.; Siu, S.; Huard, J.N.; Conner, K.P.; Milburn, R.R.; O’Neill, J.W.; Tometsko, M.E.; et al. Altering Antibody–Drug Conjugate Binding to the Neonatal Fc Receptor Impacts Efficacy and Tolerability. Mol. Pharm. 2016, 13, 2387–2396. [Google Scholar] [CrossRef]
- Li, X.; Zhou, S.; Abrahams, C.L.; Krimm, S.; Smith, J.; Bajjuri, K.; Stephenson, H.T.; Henningsen, R.; Hanson, J.; Heibeck, T.H.; et al. Discovery of STRO-002, a Novel Homogeneous ADC Targeting Folate Receptor Alpha, for the Treatment of Ovarian and Endometrial Cancers. Mol. Cancer Ther. 2022, 22, 155–167. [Google Scholar] [CrossRef]





| aCD74 ADC | ADC-X | ||||
|---|---|---|---|---|---|
| Analytical SEC | % HMW | 1% | 1% | 1% | 2% |
| % Monomer | 95% | 98% | 96% | 95% | |
| % LMW | 4% | 1% | 3% | 3% | |
| MALDI-TOF MS | DAR | 1.9 | 1.9 | 3.9 | 3.9 |
| % conj | 95% | 95% | 98% | 98% | |
| Sample | Rmax | kon (M−1s−1) | koff (M−1) | KD (M) |
|---|---|---|---|---|
| aFolR (HC/LC) | 2.4 | 4.3 × 105 | 1.2 × 10−3 | 2.8 × 10−9 |
| aFolR (PFLC) | 2.4 | 3.7 × 105 | 1.2 × 10−3 | 3.2 × 10−9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanson, J.; Groff, D.; Carlos, A.; Usman, H.; Fong, K.; Yu, A.; Armstrong, S.; Dwyer, A.; Masikat, M.R.; Yuan, D.; et al. An Integrated In Vivo/In Vitro Protein Production Platform for Site-Specific Antibody Drug Conjugates. Bioengineering 2023, 10, 304. https://doi.org/10.3390/bioengineering10030304
Hanson J, Groff D, Carlos A, Usman H, Fong K, Yu A, Armstrong S, Dwyer A, Masikat MR, Yuan D, et al. An Integrated In Vivo/In Vitro Protein Production Platform for Site-Specific Antibody Drug Conjugates. Bioengineering. 2023; 10(3):304. https://doi.org/10.3390/bioengineering10030304
Chicago/Turabian StyleHanson, Jeffrey, Dan Groff, Abi Carlos, Hans Usman, Kevin Fong, Abigail Yu, Stephanie Armstrong, Allison Dwyer, Mary Rose Masikat, Dawei Yuan, and et al. 2023. "An Integrated In Vivo/In Vitro Protein Production Platform for Site-Specific Antibody Drug Conjugates" Bioengineering 10, no. 3: 304. https://doi.org/10.3390/bioengineering10030304
APA StyleHanson, J., Groff, D., Carlos, A., Usman, H., Fong, K., Yu, A., Armstrong, S., Dwyer, A., Masikat, M. R., Yuan, D., Tran, C., Heibeck, T., Zawada, J., Chen, R., Hallam, T., & Yin, G. (2023). An Integrated In Vivo/In Vitro Protein Production Platform for Site-Specific Antibody Drug Conjugates. Bioengineering, 10(3), 304. https://doi.org/10.3390/bioengineering10030304