1. Introduction
Water is essential for all life on earth and a valuable resource for human civilization. Ensuring the consistent and dependable availability of safe and affordable water continues to be a significant worldwide challenge in the 21st century. According to the World Health Organization (WHO), over 2 billion people lack access to safe drinking water at home [
1]. Therefore, ensuring access to safe drinking water is crucial for public health and development. Per- and polyfluoroalkyl substances (PFASs), also known as “forever chemicals,” include a group of least 4700 synthetic chemicals used in various industrial and consumer applications, including food packaging, household items, and firefighting foam, since the 1940s [
2,
3] and are some the most challenging chemicals negatively impacting drinking water. PFASs consist primarily of fluorinated carbon chains (C–F). The atoms of fluorine and carbon in this bond have low polarity and high bond energy, reaching up to 536 kJ/mol [
4]. Fluorinated carbon chains are additionally functionalized by a wide range of functional groups (e.g., carboxylic acids, sulfonic acids, alcohols, etc.) located at the ends of the carbon chains. PFAS molecules can exhibit either long or short chain configurations based on the length of the carbon (C) backbone in each molecule. Examples of long-chain PFASs include but are not limited to perfluoroalkyl sulfonic acids (PFSAs) containing six or more carbon atoms and perfluoroalkyl carboxylic acids (PFCAs) containing seven or more carbon atoms. Conversely, short-chain PFASs include perfluorinated carboxylic acids (PFCAs) containing fewer than seven carbon atoms and perfluorosulfonic acids (PFSAs) containing fewer than six carbon atoms. Most of long-chain PFASs have been discontinued and replaced by short-chain PFASs. Therefore, long-chain PFASs have been occasionally referred to as legacy PFASs [
5].
PFASs have both hydrophilic and hydrophobic characteristics [
6] and high stability when exposed to thermal, biological, and chemical processes [
5,
6]. Over the past few decades, PFASs have been detected in drinking water at concentrations ranging from a few nanograms per liter (ng/L) to several hundred nanograms per liter due to different industrial and commercial activities [
4,
7,
8,
9]. These chemicals are considered dangerous to human health, even at very low concentrations, and are often measured in parts per trillion [
8,
10]. In particular, PFASs remain in the environment for decades and can cause various health issues, including kidney cancer, testicular cancer, thyroid problems, ulcerative colitis, high cholesterol, pregnancy-induced high blood pressure, hormone secretion, immune system disorder, and low birth weight in infants [
11,
12]. To reduce the likelihood of PFAS exposure through drinking water, on 10 April 2024, the U.S. EPA announced the final National Primary Drinking Water Regulation (NPDWR) for six PFAS compounds including perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), hexafluoropropylene oxide dimer acid (HFDO-DA or GenX), perfluorobutane sulfonic acid (PFBS), perfluorooctane sulfonic acid (PFOS), and perfluorohexane sulfonic acid (PFHxS). The U.S. EPA recommended that the maximum concentration levels (MCLs) of PFOA and PFOS in drinking water should not exceed 4 ng/L, while the MCLs for PFHxS, PFNA, PFBS, and GenX should not exceed 10 ng/L [
13]. In the United States, 6% to 10% of the 66,000 public drinking water systems may need to take action to meet these standards [
13]. Unfortunately, traditional technologies adopted by water treatment facilities showed a limited ability to remove PFASs [
14,
15].
The primary goal of PFAS removal is to defluorinate C–F bonding, a process hindered by the refractory and hydrophobic nature of C–F bonding, making PFASs resistant to most technologies [
16]. After treatment, long-chain PFASs can break into short-chain PFASs, which can be significantly more challenging to remove. Therefore, to remove both long- and short-chain PFASs, advanced systems (e.g., advanced oxidation process (AOP), thermal, plasma, etc.) are required. However, the implementation of these treatments significantly impacts their scale-up feasibility [
17,
18]. However, adsorption treatments (e.g., carbon nanotubes (CNT), biochar, activated carbon, etc.) represent a practical and feasible approach [
8]. For example, biochar, while effective, may require substantial amounts to treat large volumes of water. Activated carbon, although widely used, can be expensive to regenerate or replace and may not effectively remove PFBA and PFBS [
19]. These treatments offer effective PFAS removal but may have practical and economic limitations such as the high cost of materials (e.g., adsorption treatments), complex production processes, and challenges in large-scale application (e.g., AOP) [
20]. Among the new adsorption materials, graphene, due to its unique properties, represents a possible solution. Graphene is a single layer of carbon atoms arranged in a two-dimensional honeycomb lattice, possessing exceptional strength, conductivity, and flexibility, making it one of the most promising materials for various technological applications. Additionally, graphene can be easily functionalized and has demonstrated remarkable efficacy in the removal of many environmental contaminants, including heavy metals, organic pollutants, and microorganisms from water [
21,
22,
23].
PFASs are persistent environmental pollutants increasingly detected in water sources, posing serious health and ecological risks due to their chemical stability and resistance to degradation. Despite growing regulatory attention, the effective removal of PFASs from water remains a major challenge, largely due to their strong carbon–fluorine bonds and varied chemical structures [
9]. While previous studies have explored the use of carbon-based adsorbents for PFAS remediation, many have focused on limited compound types or generalized PFAS behavior without accounting for the structural diversity among individual species [
4]. This study aims to address this gap by systematically evaluating graphene’s potential as an adsorbent for six PFAS compounds currently regulated by the U.S. EPA: PFOA, PFNA, GenX, PFBS, PFOS, and PFHxS. Molecular dynamics (MDs) simulations were employed to investigate the adsorption behavior of these compounds individually and in a multicomponent system. It is important to note that adsorption at the molecular scale can be modeled using various simulation techniques. More rigorous approaches, such as Grand Canonical Monte Carlo (GCMC) simulations and molecular dynamics-based umbrella sampling, enable direct sampling of adsorption processes and facilitate the calculation of adsorption free energies or isotherms by explicitly capturing the exchange between bulk and adsorbed phases [
24,
25]. However, due to their high computational cost, these methods are often complemented by conventional molecular dynamics simulations that focus on evaluating relative interaction energies and structural trends, which can still provide meaningful insights for screening adsorbent materials [
26]. Additionally, it introduces novel analyses, including diffusion coefficient calculations and the measurements of distances between graphene and the head and tail groups of each PFAS molecule—approaches not previously documented in the literature, to the best of our knowledge.
2. Computational Method
2.1. Molecular Models Compounds and Structures
The molecular models used in this study included graphene, water, and six per- and polyfluoroalkyl substances (PFASs): GenX, PFBS, PFNA, PFOA, PFHxS, and PFOS. These models were developed using Biovia Materials Studio® (Version 2022).
A finite hexagonal lattice of carbon atoms was constructed to represent graphene, with the chemical formula C
86H
48 and a lattice dimension of 15 × 15 Å. This size was chosen to be 1.5 times the dimensions of PFAS molecules to ensure sufficient interaction space, avoid edge effects, and allow full surface interaction during adsorption simulations, following the methodology described in [
27].
Figure 1 illustrates the modeled graphene sheet.
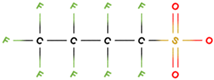
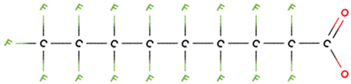
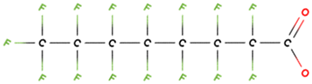
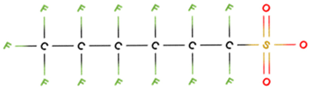
PFAS molecular structures vary based on their head groups. PFOA, PFNA, and GenX possess a carboxylate head, whereas PFBS, PFOS, and PFHxS feature a sulfonate head. Among the carboxylate-terminated PFASs, PFOA and PFNA contain eight and nine fluorinated carbon atoms, respectively, while GenX consists of a hexafluoropropylene oxide dimer with an acid functional group and six carbon atoms. In contrast, the sulfonate-terminated PFASs include PFBS, PFHxS, and PFOS, which contain four, six, and eight fluorinated carbon atoms, respectively.
This structural distinction between carboxylate and sulfonate head groups plays a crucial role in the adsorption behavior and interaction strength with graphene, as explored in subsequent sections. The chemical properties of the PFAS compounds are presented in
Table 1, with their molecular structures visualized using the open-source software MolView (Web Version, Updated August 2024).
It is important to note that PFASs are predominantly found in their anionic (negatively charged) state under natural aqueous conditions, whereas neutral PFASs typically occur in environments with pH levels below 3.7 [
28]. Given the relevance of environmental conditions to real-world water purification scenarios, this study specifically focused on the behavior of anionic PFASs.
2.2. Molecular Simulation Details
Geometry optimization is a computational process used to identify the most stable structure of a molecule or molecular system by adjusting the positions of its atoms. The goal is to minimize the potential energy of the system, leading to a configuration where the forces acting on each atom are as close to zero as possible. This process involves iteratively moving the atoms according to calculated forces and potential energy gradients until a stable, low-energy configuration is achieved [
29]. Geometry optimization was conducted on each PFAS compound and on the graphene sheet before constructing the amorphous cell. In this study, the amorphous cell was constructed with six graphene sheets (15 × 15 Å), ten PFAS molecules, ten sodium atoms, and a thousand water molecules for each PFAS compound. The NPT (isothermal–isobaric ensemble) approach was used to enable the specification of the number of molecules, pressure, and temperature in the cell, while the program determined and calculated the volume and energy of the system. The pressure was set at 1.013 × 10
−4 GPa, while the temperature value was set at 298 K in accordance with previously reported values [
8,
20,
30]. Temperature and pressure were controlled using standard algorithms implemented within BIOVIA Materials Studio. A time step of 1 fs was used, with sufficient equilibration and production run durations to ensure system stability.
Figure 2 highlights the amorphous cell constructed for each PFOA compound after applying the geometry optimization and the NPT approach.
In MD simulations, a force field represents a mathematical representation of the potential energy associated with a particle system. It characterizes bonded (e.g., bond stretching, angle bending, and dihedral angles) and non-bonded (e.g., van der Waals forces and electrostatic interactions) interactions and effects that the particles exert on each other’s motion over time. The COMPASS force field was selected because it is well-suited for condensed-phase simulations and accurately models both covalent and non-covalent interactions, including van der Waals forces, hydrogen bonding, and electrostatic interactions. This makes it particularly effective for studying the adsorption behavior of fluorinated compounds like PFAS on surfaces such as graphene. The COMPASS framework is also designed to replicate the physical properties of both individual molecules and their aggregated states, making it a suitable choice for modeling PFAS–graphene interactions in aqueous environments.
After testing different simulation periods (e.g., 3.0, 4.0, 5.0, and 6.0 ns with a time step of 1 fs) with the COMPASS forcefield, a period of 3.0 ns was selected due to the system reaching an equilibrium state. In addition to testing the interaction between each PFAS and graphene, to mimic the occurrence of several PFASs in water, all six PFASs investigated were placed together in a single amorphous cell. Overall, two molecules of each PFAS (GenX, PFBS, PFNA, PFOA, PFHxS, and PFOS), six graphene sheets, twelve sodium atoms, and a thousand water molecules were simultaneously added. After that, the adsorption energy for each PFAS type under these competitive conditions was calculated. It should be noted that due to computational constraints, each simulation in this study was performed once with no replicates for the single and the multicomponent PFAS compound. However, two verification simulations were conducted, and the resulting adsorption energies showed minimal variation, supporting the reliability of a single simulation for all compounds.
2.3. Adsorption Energy and Diffusion Coefficient
The adsorption energy (
Ead) and the diffusion coefficient (
D) are essential to better understand the interaction between graphene and PFAS.
Ead is defined as the sum of the electrostatic and van der Waals interactions between the molecules (specifically, between the PFAS and graphene) throughout the simulation and averaged over each step. Also, it refers to the binding affinity of one molecule to another. The trajectories and the energy components of the systems were obtained and analyzed every 100 ps over the last 500 ps to calculate the various interaction energies using Equation (1) [
30].
where
Ead: adsorption energy;
Esys: total system energy (graphene + PFAS); and
E1 and
E2: energies of the individual components, namely graphene and PFAS, respectively.
The diffusion coefficient quantifies the rate at which particles diffuse through a medium, reflecting the ease of movement from high to low concentration regions. It is measured in units of area per time. To determine the diffusion coefficient, the mean-square displacement (
MSD) of the atoms was computed by analyzing the time series of all atomic locations (
r) using the following formula [
30].
where
MSD: mean-square displacement;
t: simulation time.
Then, the Einstein relation was used for a three-dimensional system as follows to calculate the diffusion coefficient.
2.4. Distance Measurement Analysis of PFASs Using Graphene
The distances between the center of the graphene sheet and the head and tail groups of each PFAS compound were measured to provide additional insights into their adsorption behavior. By analyzing these distances, it is possible to assess whether specific structural features, such as the functional head group (sulfonate or carboxylate) or the perfluorinated tail, play a more significant role in adsorption. Additionally, these measurements allow for the correlation between molecular distances and adsorption energy to be calculated as shorter distances indicate stronger interactions and enhanced PFAS capture by graphene. This analysis aims to further elucidate the relationship between adsorption affinity and the structural characteristics of PFAS compounds.
2.5. Statistical Analysis
To evaluate the impact of molecular weight on PFAS adsorption energy, an independent samples
t-test was performed using JASP [
31,
32] with a significance threshold of
p-value < 0.05. A
t-test is a statistical method used to determine whether there is a significant difference between the means of two independent groups. In this case, PFAS compounds were categorized into low and high molecular weight groups.
The t-test is essential for this analysis because it allows for the objective statistical evaluation of whether molecular weight has a meaningful effect on adsorption energy, beyond what might be expected by random variation. By confirming that the observed differences are statistically significant, this test strengthens the reliability of the simulation results and supports the conclusion that molecular weight plays a critical role in PFAS adsorption behavior.
3. Results and Discussion
3.1. Adsorption Energy of Individual PFAS
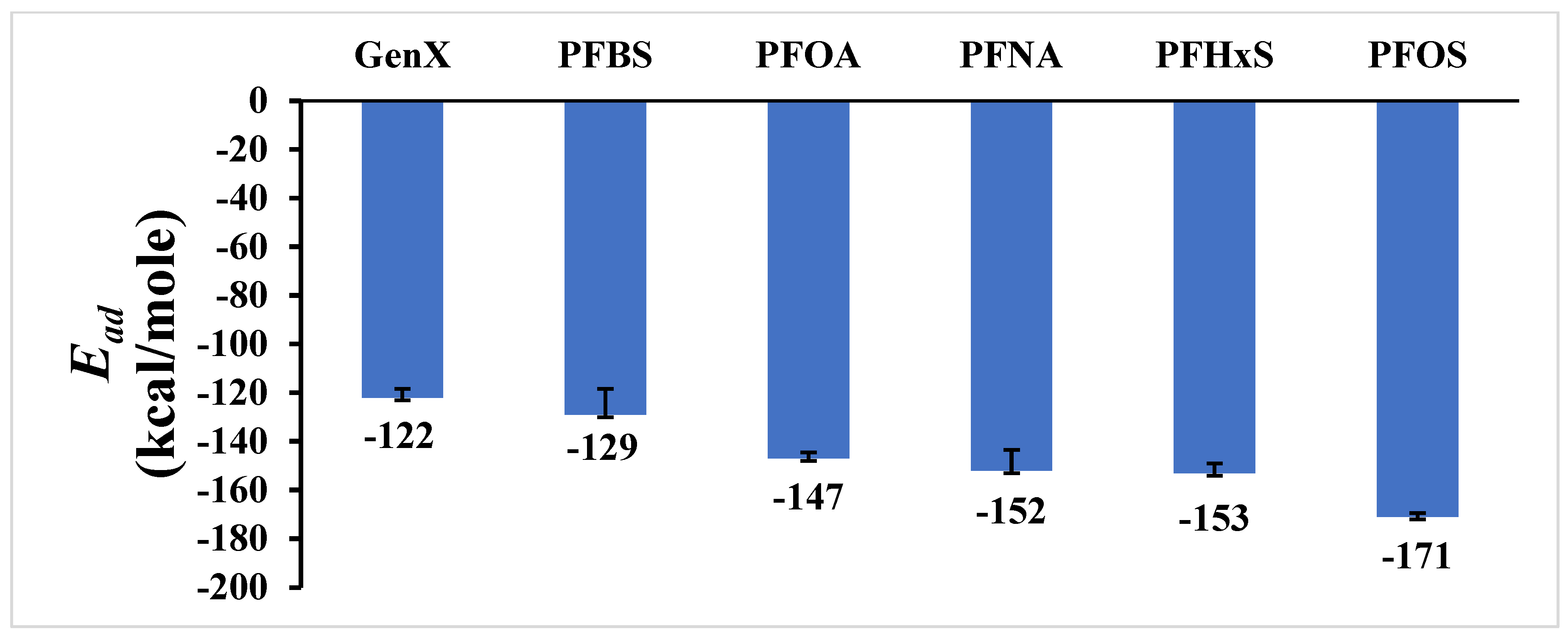
Table 2 presents the adsorption energy calculations for the PFAS compounds, and also the van der Waals and electrostatic energies, along with the chemical formulas and molecular weights of the PFAS compounds.
Figure 3 provides a schematic representation of the adsorption energy results.
Among the six investigated PFAS molecular structures, PFOS had the highest molecular weight (500 g/mol) as well as the most negative adsorption energy of −171 kcal/mole, indicating the highest interaction rate with graphene (
Table 1 and
Figure 3). This suggests a strong correlation between higher molecular weight and stronger adsorption (
p-value < 0.001). Following PFOS, PFHxS, which has a molecular weight of 400 g/mol, exhibits an adsorption energy of −153 kcal/mole, also indicating a robust interaction with graphene. PFNA and PFOA show adsorption energies of −152 kcal/mole and −147 kcal/mole, respectively, with molecular weights of 464 g/mol and 414 g/mol. These results further support the trend that higher molecular weights correlate with enhanced adsorption energies. This was also confirmed by performing a statistical analysis using JASP software, which provided a
p-value of less than 0.001. PFBS and GenX exhibit the least negative adsorption energies of −129 kcal/mole and −122 kcal/mole, respectively, indicating weaker interactions with graphene. PFBS has the lowest molecular weight of 300 g/mol, followed by GenX (330 g/mol), reinforcing the observation that lower molecular weights tend to result in weaker adsorption. Additionally, the number of carbon atoms in the PFAS compounds also affects the adsorption energy, with the general trend that more carbon atoms enhance the interaction with graphene. PFOS and PFNA, with eight and nine carbon atoms, respectively, showed strong adsorption energies, while PFBS and GenX, with four and six carbon atoms, respectively, showed weak adsorption energies. Furthermore, the type of head sulfonic acid group used versus carboxylate also influenced the adsorption energy. Compounds with sulfonic acid head groups (PFOS, PFHxS, and PFBS) generally exhibited stronger adsorption energies compared to those with carboxylate head groups (PFOA, PFNA, and GenX). PFOS and PFHxS, both with sulfonic acid head groups, had some of the most negative adsorption energies at −171 kcal/mole and −153 kcal/mole, respectively, indicating easier adsorption onto graphene. By comparing the influence of the head group versus the number of carbon atoms, the head group had a more dominant effect on adsorption. This is evidenced by PFOS and PFHxS having stronger adsorption energies than some compounds with more carbon atoms but different head groups. While the number of carbon atoms enhances adsorption to some extent, the type of head group, particularly the presence of sulfonic acid, plays a more critical role in determining the strength of adsorption. Thus, the analysis reveals that molecular weight, the number of carbon atoms, and the type of head group all play crucial roles in the adsorption of PFAS compounds onto graphene. However, the head group chemistry, especially the sulfonic acid head group, is more dominant in enhancing adsorption energy compared to the number of carbon atoms and molecular weight, suggesting that PFOS and PFHxS, due to their significant molecular weights, higher number of carbon atoms, and favorable head group chemistry, exhibit the strongest adsorption affinities for graphene. These results are in line with previously published results with enhanced adsorption for long-chain PFASs and for those with sulfonic head groups rather than those with shorter chains or with carboxyl head groups [
20,
33,
34]. Similar results were also observed in [
20], where the authors investigated the adsorption mechanism of three different types of PFAS, PFBA, PFOA, and PFOS, in their anionic state in the presence of graphene and water. It was found that PFASs with sulfonic head (PFOS) had the highest adsorption energy compared to the other two PFAS compounds with hydroxylate head. Additionally, PFAS compounds with higher chain length (PFOS and PFOA) had higher amounts of adsorption energy compared to those with shorter chain lengths (PFBA) [
20]. Jiang et al., 2021, investigating three different PFAS compounds (PFOS, PFOA, and PFBA) with sulfonic head, observed that the PFAS compounds with longer chain length provided the highest adsorption energy [
8].
3.2. Adsorption Energy of Multicomponent PFAS System
Table 3 presents the adsorption energy calculations, as well as the
EvdW and
Es, for the PFAS compounds in the multicomponent PFAS system, along with their chemical formulas.
Figure 4 provides a schematic representation of the adsorption energy.
The adsorption behavior of the PFAS compounds on graphene significantly changed when they occurred simultaneously in a multicomponent PFAS system (
Table 3 and
Figure 4 vs.
Table 2 and
Figure 3—PFASs individually added). The adsorption energies for all selected PFASs were less negative in the multicomponent PFAS system, indicating weaker interactions with graphene due to competitive adsorption. The previously observed relationship between larger molecular weight and stronger adsorption in the individual system was less prominent in the multicomponent PFAS system. The PFOS compound with a molecular weight of 500 g/mole and the highest adsorption energy in the individual system, exhibits a comparatively lower adsorption energy of −16 kcal/mol in the multicomponent PFAS system compared to other PFAS compounds such as PFOA and GenX. PFOA and GenX, with molecular weights of 414 g/mole and 350 g/mole, respectively, showed the highest adsorption energies of −32 kcal/mol each in the multicomponent PFAS system.
The influence of the head groups was also altered in the multicomponent PFAS system, with PFOS and PFHxS (sulfonic acid head groups) showing slightly weaker adsorption energies (−16 and −28 kcal/mol, respectively) compared to PFOA and GenX (carboxylate head groups, −32 kcal/mol for both), suggesting that competition for adsorption sites diminishes the dominance of sulfonic acid head groups. This competition reduces the adsorption efficiency, resulting in less negative adsorption energies, as PFAS molecules with stronger individual adsorption energies are hindered by the presence of other PFAS types, leading to a more uniform distribution of adsorption energies. PFOA and GenX exhibit the most negative adsorption energies, indicating relatively stronger interactions with graphene, while PFBS and PFOS show the least negative adsorption energy (−16 kcal/mol for both), suggesting the weakest interaction. These findings highlighted the importance of considering collective behavior in the multicomponent PFAS system for practical applications involving the PFAS multicomponent PFAS system and graphene.
It is also noted that in the multicomponent PFAS system, adsorption energies were generally less negative compared to the individual systems, indicating weaker interactions due to competitive adsorption among PFAS compounds for graphene’s active sites. This suggests that in real-world water contamination scenarios, where multiple PFASs are present simultaneously, the adsorption performance of graphene may be reduced. Notably, PFOS, which showed the strongest adsorption in the individual system, exhibited significantly lower adsorption energy when introduced with other PFASs, highlighting that structural advantages like the molecular weight and head group are less influential under competitive conditions. Despite this reduction, graphene still demonstrated a consistent ability to absorb all PFAS types, making it a viable material for water purification applications.
3.3. Diffusion Coefficient Results
The diffusion coefficient results of all PFAS compounds are shown in
Table 4. The diffusion coefficients of the different PFAS compounds in water and in water with the presence of graphene revealed that graphene enhances the diffusion of PFAS compounds. For instance, GenX showed a diffusion coefficient increase from 9.67 × 10
−7 cm
2/s in water to 1.94 × 10
−6 cm
2/s with graphene, nearly doubling its mobility. PFBS exhibited an increase of more than double from 1.71 × 10
−9 cm
2/s to 3.56 × 10
−9 cm
2/s. PFNA and PFHxS also showed high increases in diffusion coefficients with graphene from 1.67 × 10
−7 cm
2/s to 2.51 × 10
−6 cm
2/s and from 3.28 × 10
−7 cm
2/s to 1.56 × 10
−6 cm
2/s, respectively. PFOA, with a slight increase from 4 × 10
−6 cm
2/s to 4.21 × 10
−6 cm
2/s, indicates moderate enhancement. PFOS showed a substantial increase from 2.5 × 10
−7 cm
2/s to 1.45 × 10
−6 cm
2/s. The molecular weight of PFAS compounds inversely affected the diffusion coefficient, with lighter molecules like PFBS having higher diffusion coefficients and heavier ones like PFOS showing lower values. Additionally, the number of carbon atoms in the PFAS compounds influenced the diffusion coefficients. Compounds with fewer carbon atoms, such as PFBS (four carbon atoms), generally have higher diffusion coefficients, while those with more carbon atoms, like PFOS (eight carbon atoms) and PFNA (nine carbon atoms), have lower diffusion coefficients. The presence of graphene generally mitigates this effect, enhancing the diffusion coefficients across all compounds and indicating improved mobility in the graphene–water medium. This enhancement in diffusion due to graphene implies that water treatment processes involving graphene can remove PFAS compounds much more quickly. Thus, while molecular weight and the number of carbon atoms influence the diffusion coefficient, the presence of graphene plays a crucial role in enhancing PFAS diffusion, suggesting that using graphene could lead to very fast and efficient treatment of water contaminated with PFAS. Bresnahan (2023) also indicated that graphene is an excellent adsorbent to PFAS contaminants [
20]. This enhancement may result from specific interactions between PFAS molecules and graphene’s hydrophobic, planar surface, which can reduce resistance to movement and potentially alter the local water structure. Such effects lower the energetic barrier to diffusion, allowing PFASs to migrate more freely. In practical terms, this means that PFASs can reach the adsorbent surface more quickly, improving adsorption kinetics.
3.4. PFAS–Graphene Distance Analysis Results of Individuals
The distances between the center of graphene and the head and tail groups of each PFAS compound exhibited a strong correlation with the previously determined adsorption energy trends, providing deeper insight into the governing adsorption mechanisms (
Table 5). Across all PFAS compounds, the tail groups are consistently positioned closer to the graphene surface than the head groups, indicating that hydrophobic interactions and van der Waals forces between the perfluorinated tail and graphene play a significant role in adsorption. PFOS, which demonstrated the most negative adsorption energy (−171 kcal/mole), exhibits one of the shortest molecular distances to graphene, with the tail group positioned at 6.15 Å and the head group at 8.23 Å, confirming its strong interaction. Similarly, PFHxS, which had the second most negative adsorption energy (−153 kcal/mole), maintained close proximity to graphene, with tail and head group distances of 6.47 Å and 8.24 Å, respectively. PFNA and PFOA, which exhibited intermediate adsorption energies (−152 kcal/mole and −147 kcal/mole, respectively), also maintained relatively short distances, though these were slightly greater than those observed for PFOS and PFHxS, reinforcing their strong but comparatively lower adsorption affinities.
Conversely, GenX and PFBS, which exhibited the least negative adsorption energies (−122 kcal/mole and −129 kcal/mole, respectively), were positioned significantly farther away from the graphene surface, with both their head and tail groups exceeding 30 Å and 45 Å. This trend further supports the relationship between weaker adsorption energies and greater molecular distances, indicating a reduced interaction with graphene. Furthermore, these findings reaffirm the fact that sulfonic acid head groups (e.g., PFOS, PFHxS, and PFBS) generally enhance adsorption relative to carboxylate head groups (e.g., PFOA, PFNA, and GenX). The shorter distances of sulfonic acid-containing PFAS, particularly PFOS and PFHxS, further highlight the importance of head group chemistry in adsorption.
In addition, the influence of chain length is evident, as longer-chain PFASs demonstrate stronger adsorption and shorter molecular distances to graphene, while shorter-chain compounds, such as PFBS, remain significantly farther from the surface. However, despite the contributions of molecular weight and chain length, the consistent observation that tail groups are positioned closer to graphene than head groups underscores the dominant role of hydrophobic interactions in adsorption.
4. Conclusions
This study’s objective was to derive the design principles for PFAS removal using molecular dynamics simulations of six EPA-regulated PFAS compounds interacting with graphene. The simulations assessed adsorption energy, diffusion coefficients, and PFAS–graphene distances, both individually and in realistic multicomponent PFAS environments with water. The findings clearly demonstrate that graphene is a highly promising material for PFAS removal in water treatment. Higher-molecular-weight PFAS compounds exhibited stronger adsorption, with PFOS showing the highest interaction strength (500 g/mol, −171 kcal/mol). Sulfonic acid-terminated PFAS, like PFOS (−171 kcal/mol) and PFHxS (−153 kcal/mol), were adhered more strongly by graphene than carboxylate-terminated counterparts such as PFNA (−152 kcal/mol) and PFOA (−147 kcal/mol). Furthermore, graphene-enhanced PFAS diffusion in water potentially accelerates contaminant removal.
In the multicomponent PFAS system, competitive adsorption slightly weakened individual adsorption energies and the molecular weight-adsorption strength correlation. Nonetheless, graphene maintained a strong overall adsorption capacity, underlining its robustness even under complex, realistic conditions. These insights reinforce graphene’s significant potential as a next-generation adsorbent, offering a strategic pathway for developing highly efficient water purification technologies targeting persistent contaminants like PFAS.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}