


Insight into the Structural and Dynamical Processes of Peptides by Means of Vibrational and Ultrasonic Relaxation Spectroscopies, Molecular Docking, and Density Functional Theory Calculations



Abstract
1. Introduction
2. Materials and Methods
2.1. Solution Preparation
2.2. Infrared and Electronic Absorption Measurements
2.3. Acoustic Absorption and Velocity Measurements
2.4. Density Functional Theory (DFT) Calculations
2.5. Molecular Docking Calculations
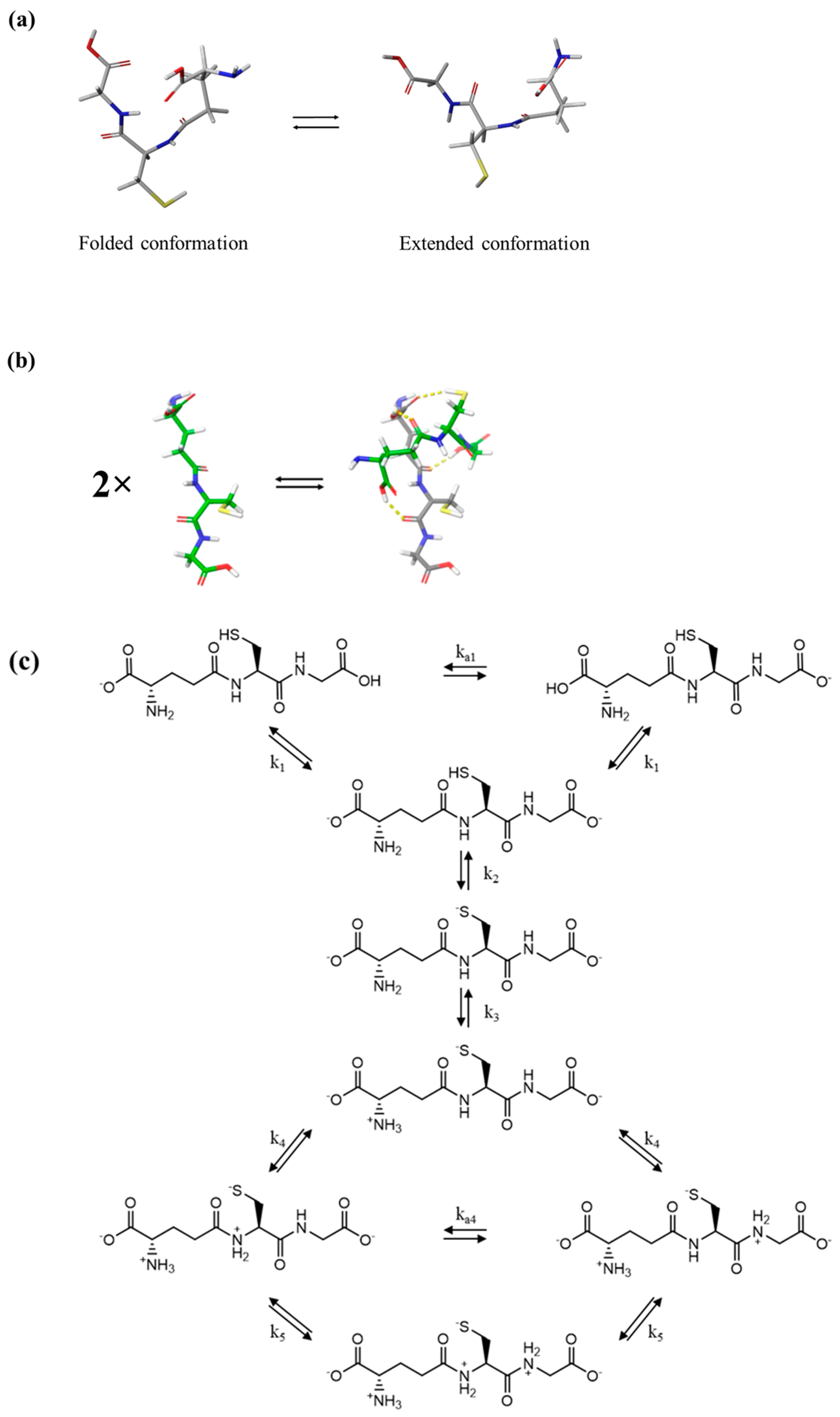
2.6. Conformational Search and Clustering Procedures
3. Results and Discussion
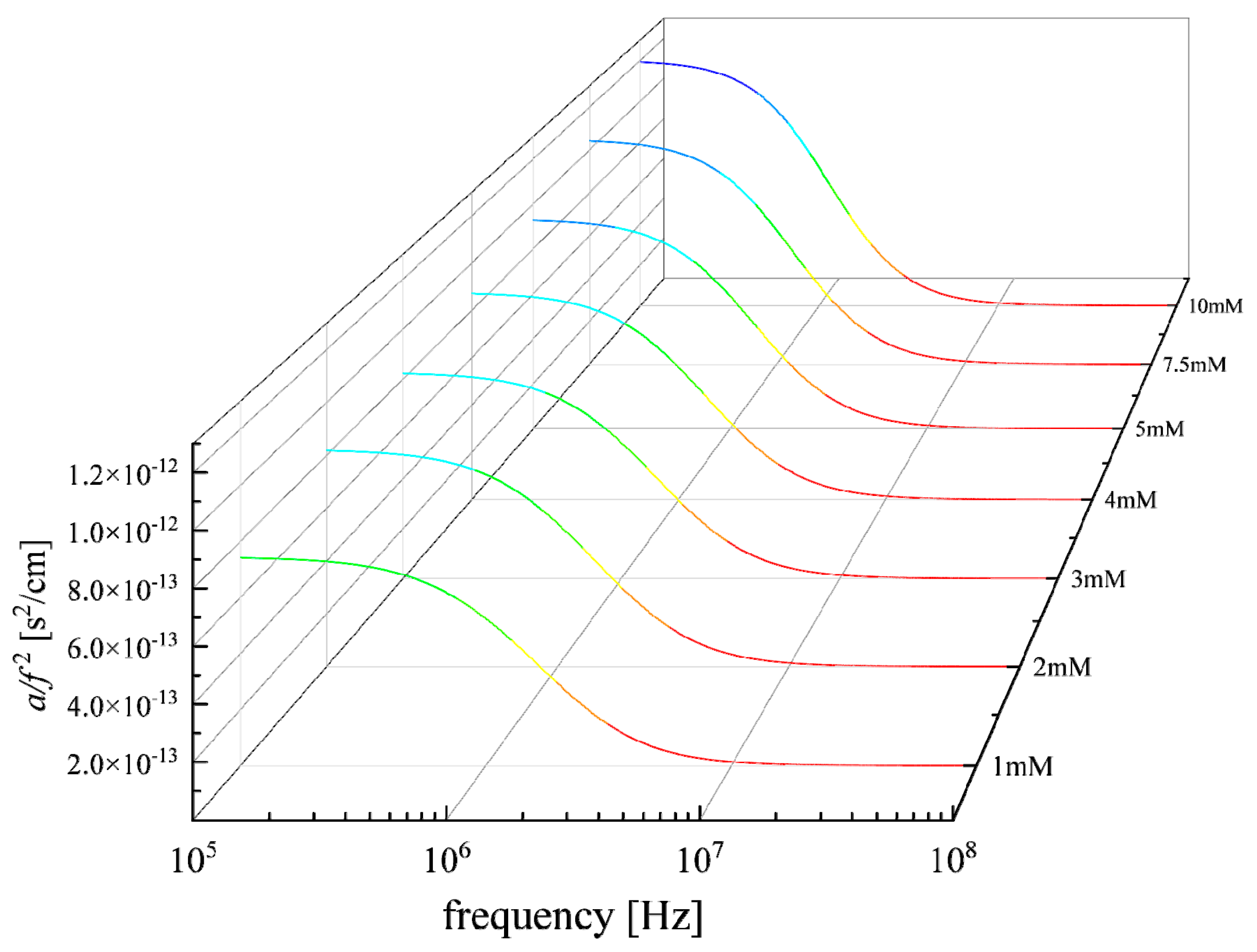
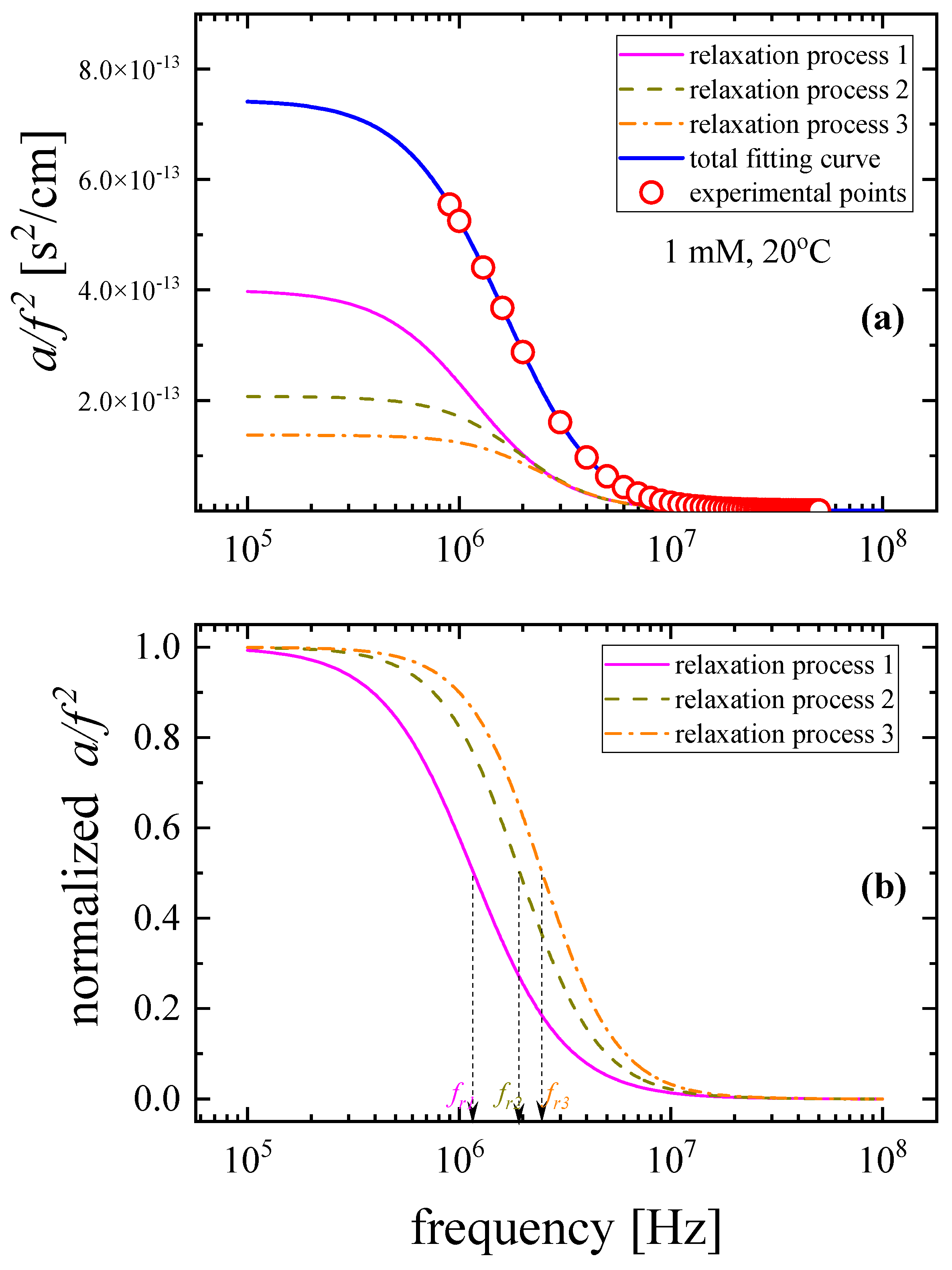
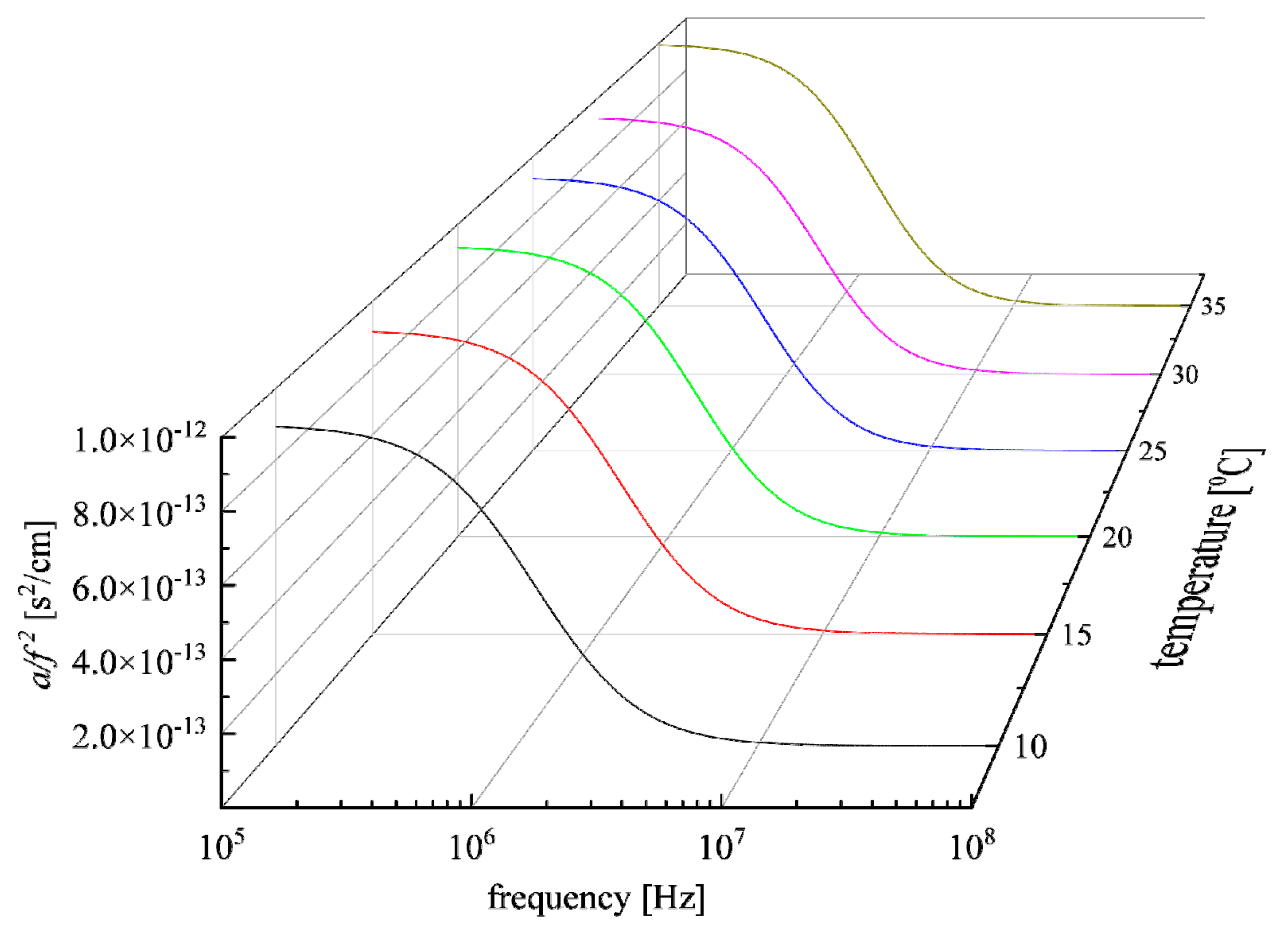
3.1. Relaxation Processes Occurring in GSH Aqueous Solutions
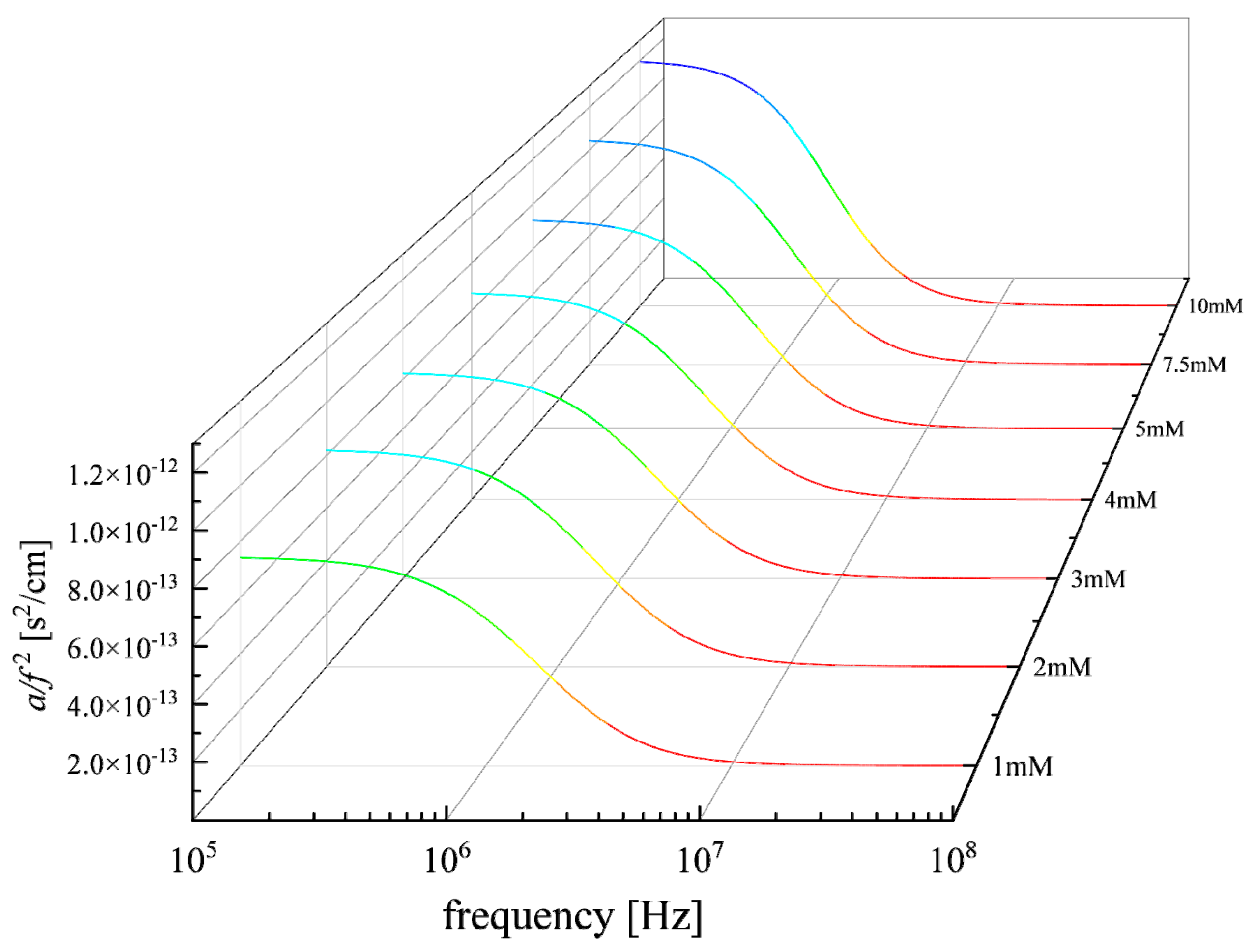
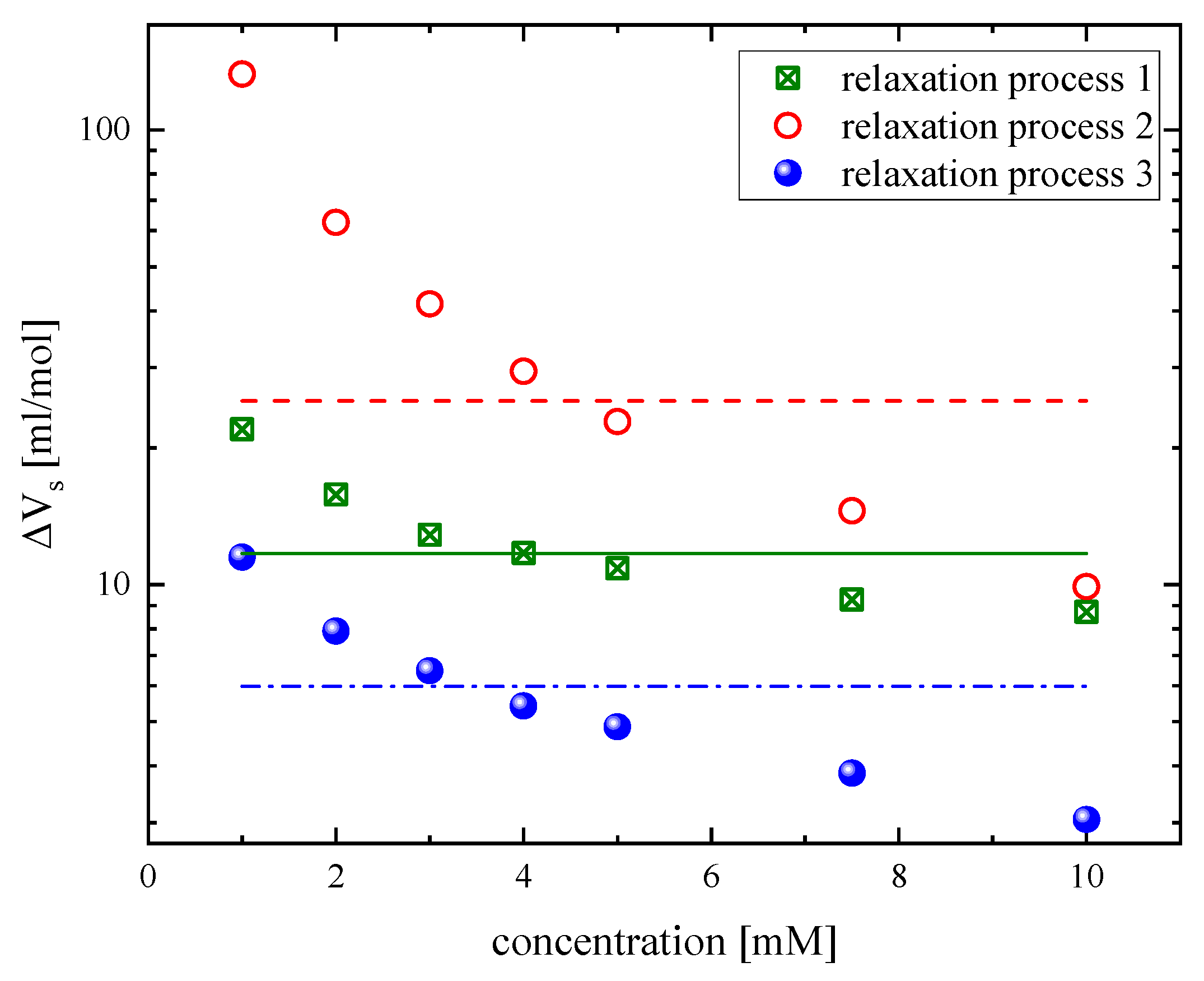
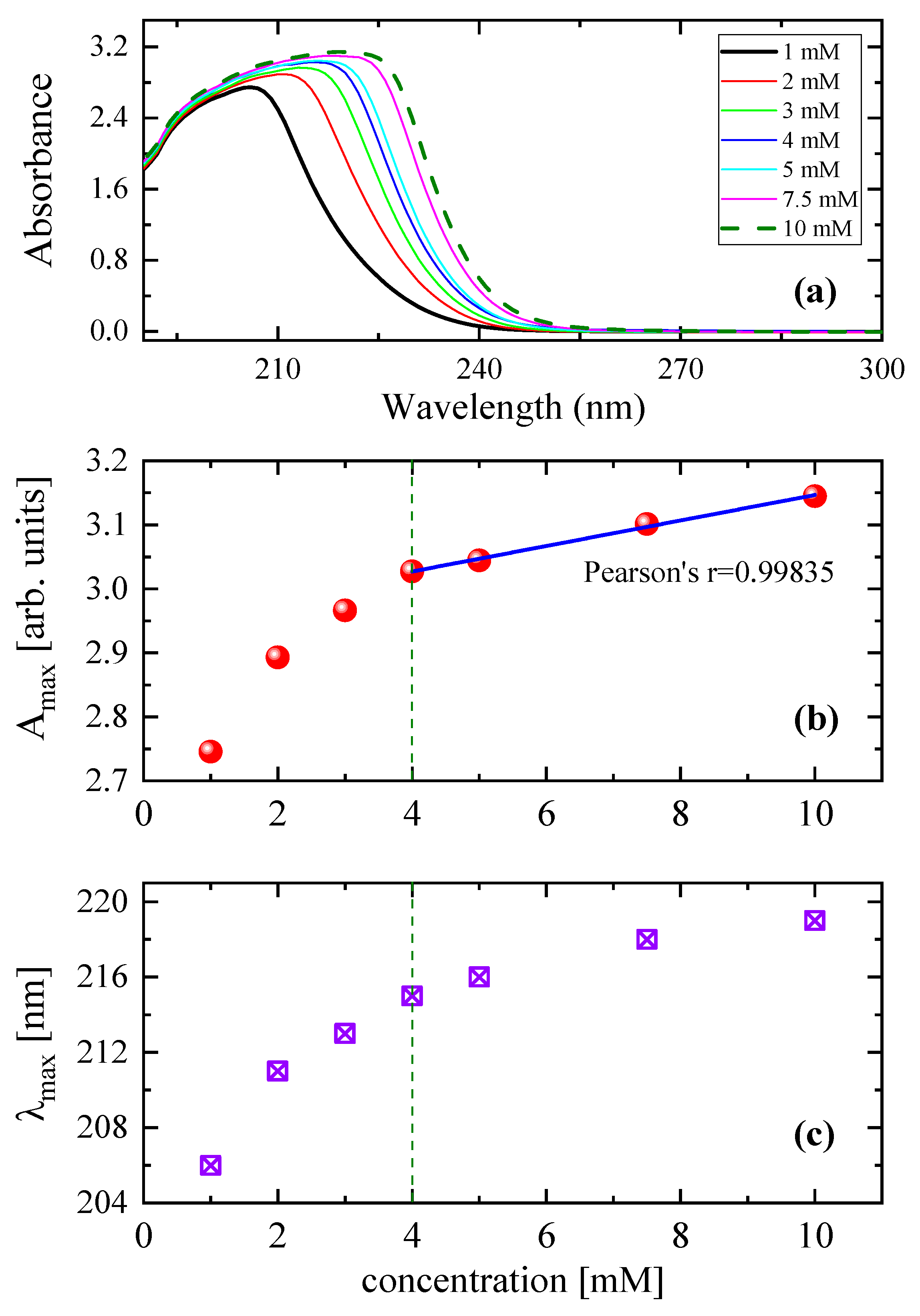
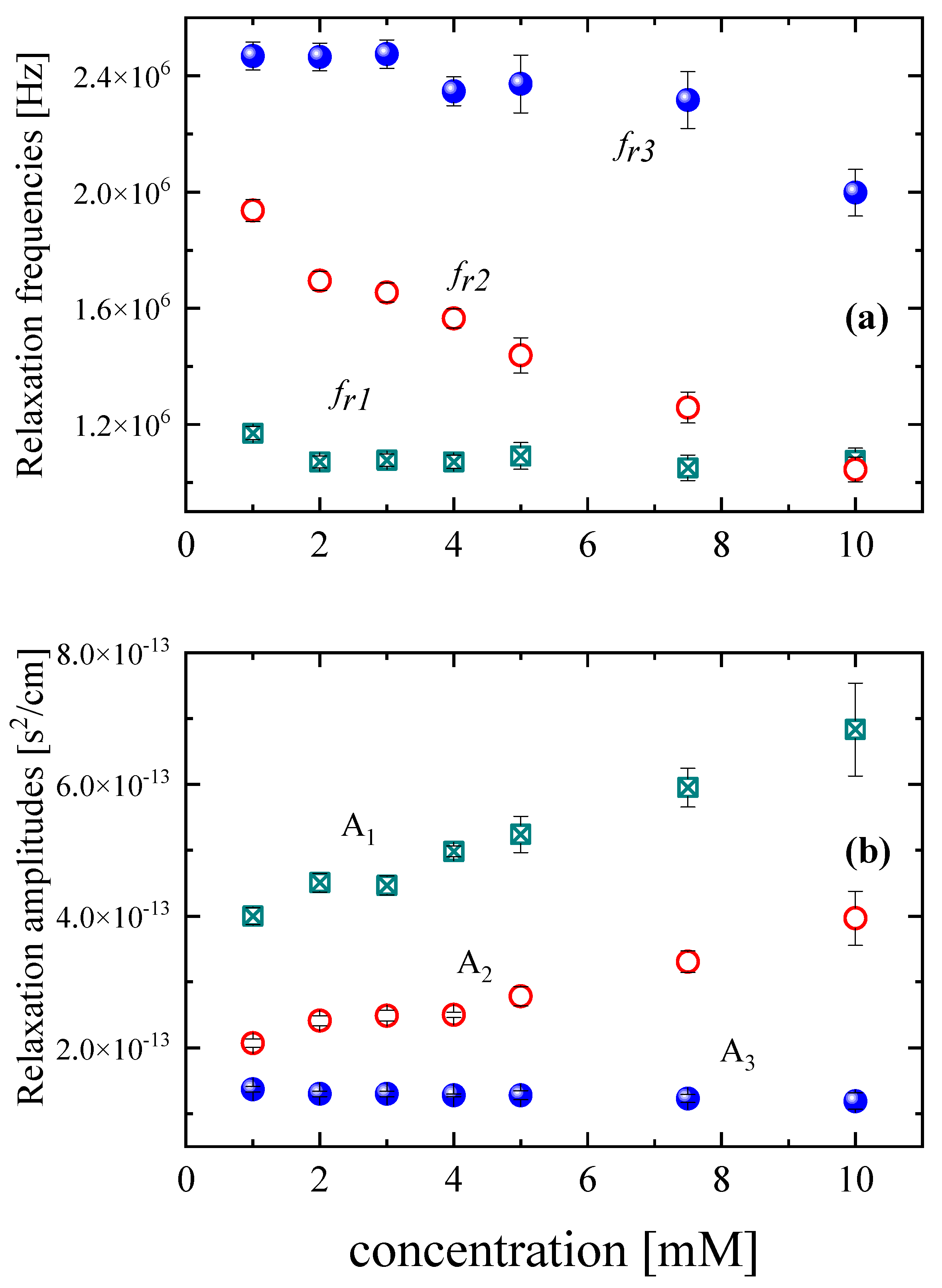
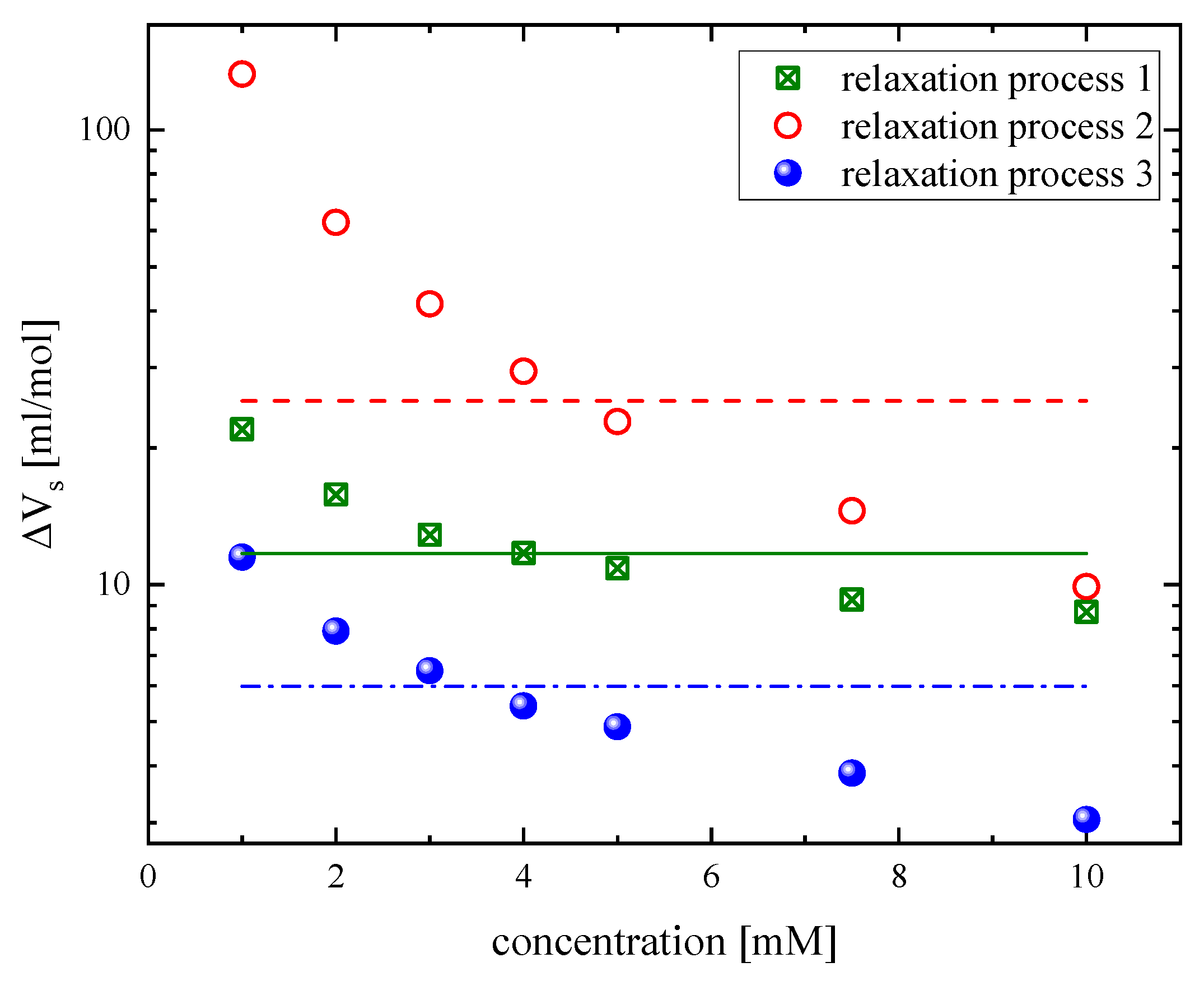
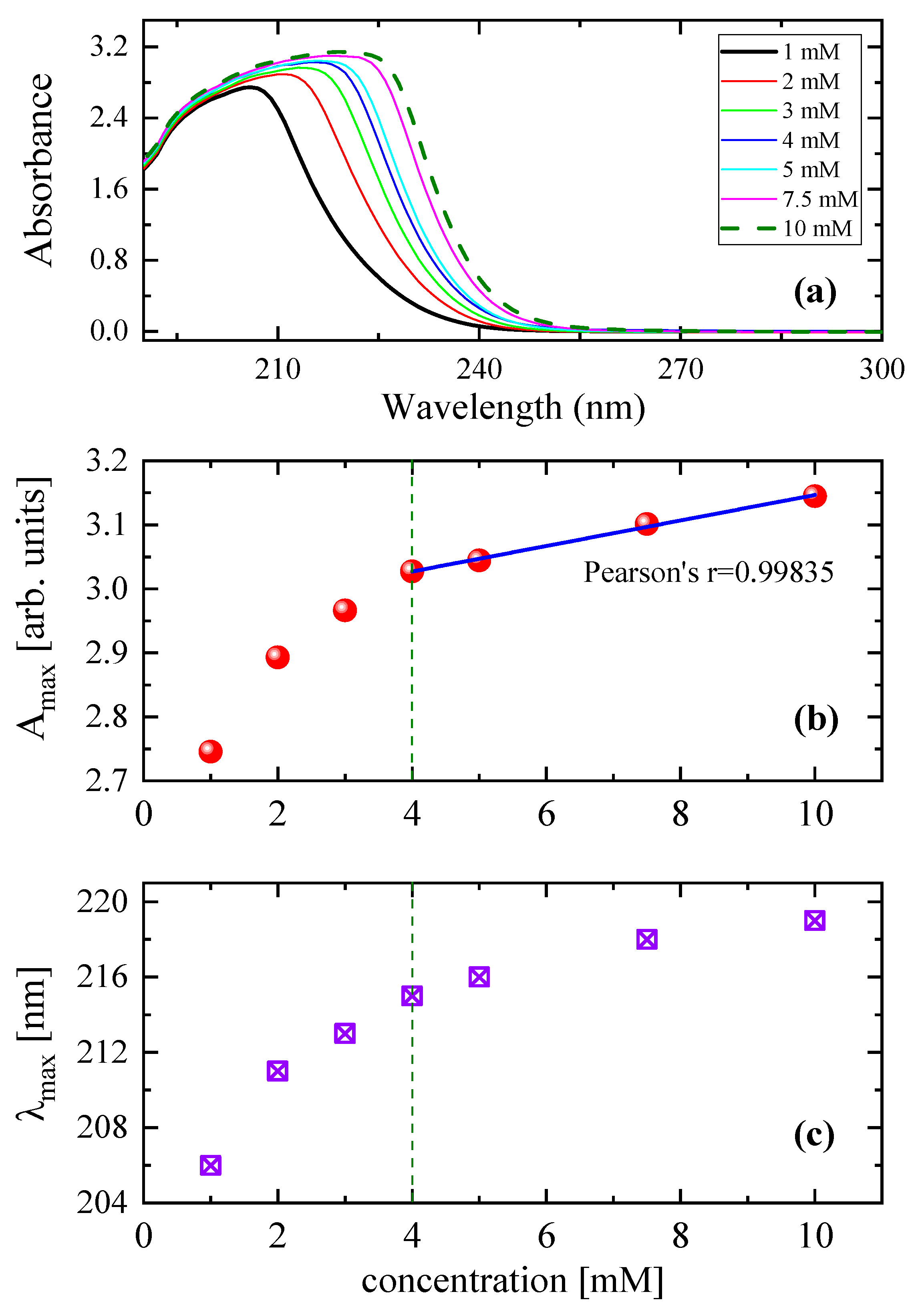
3.2. Effect of Concentration on the Relaxation Behavior
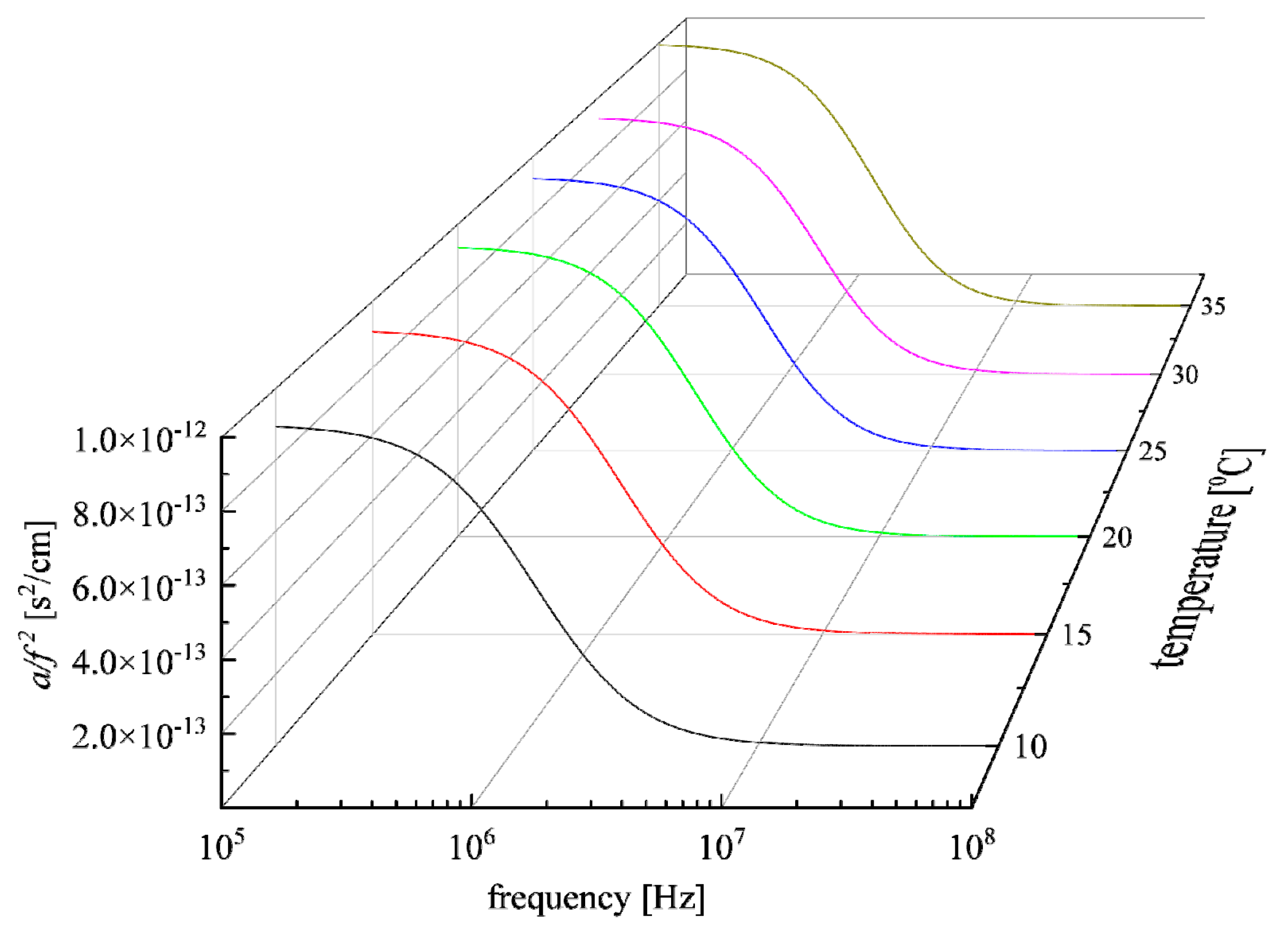
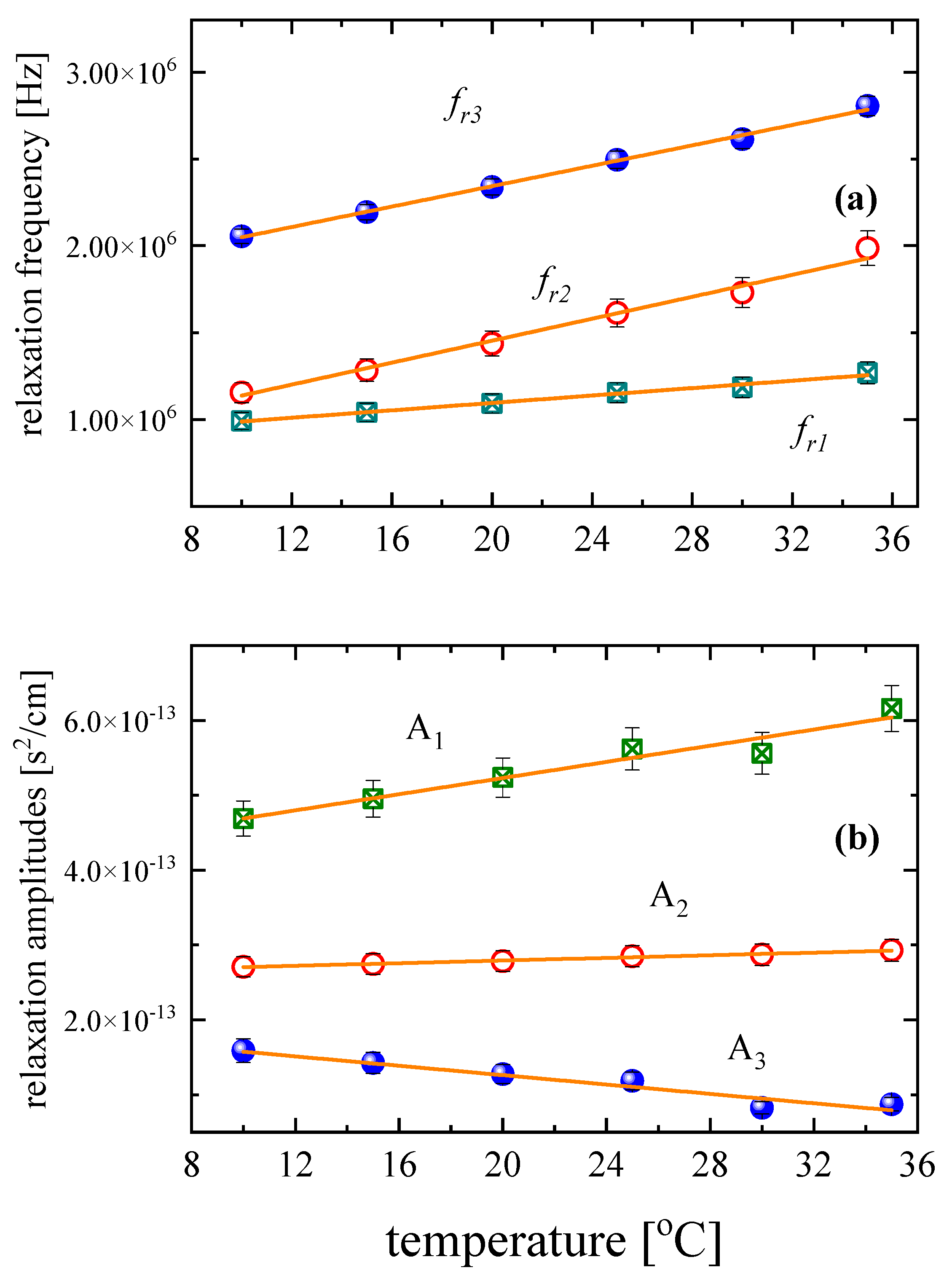
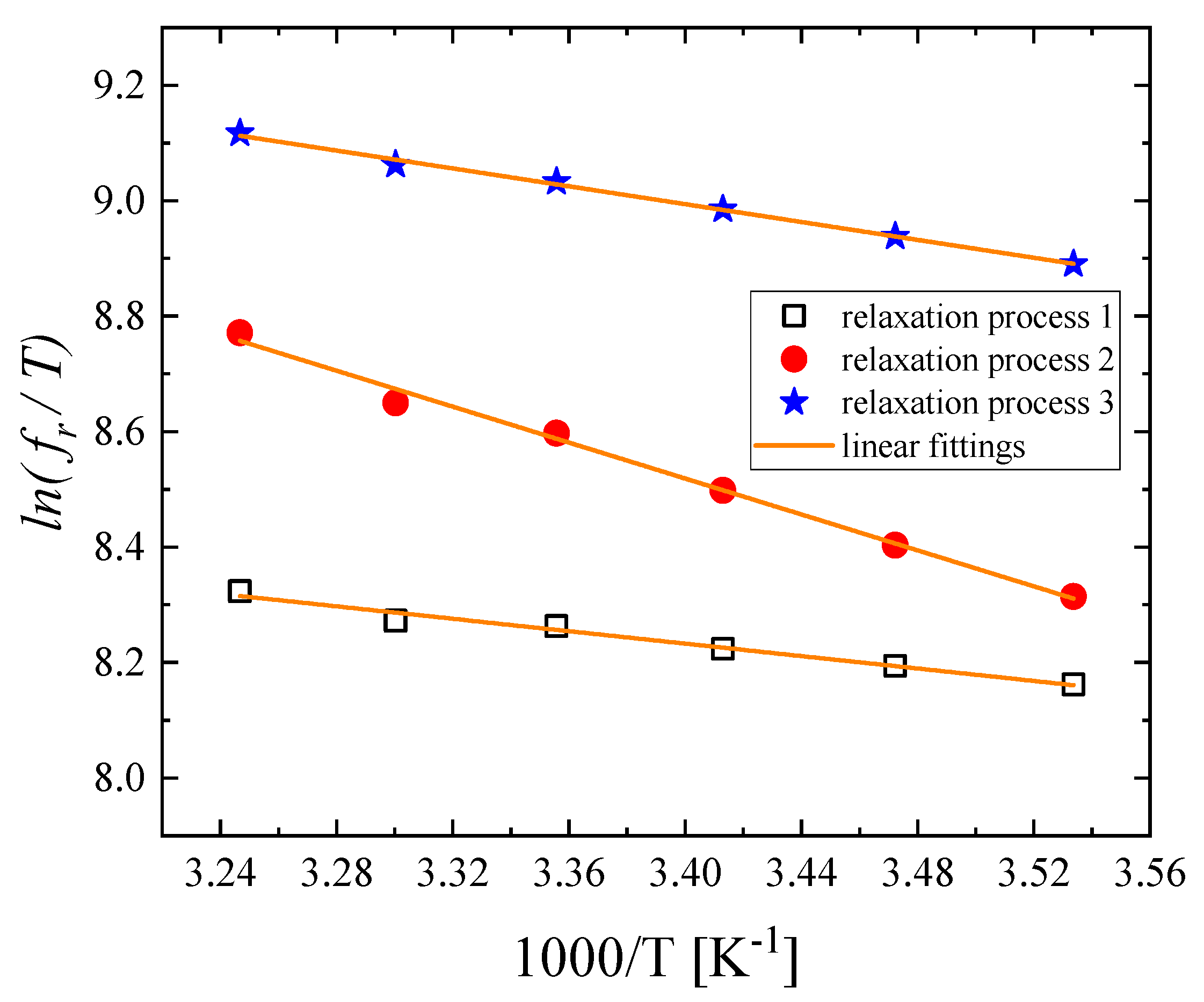
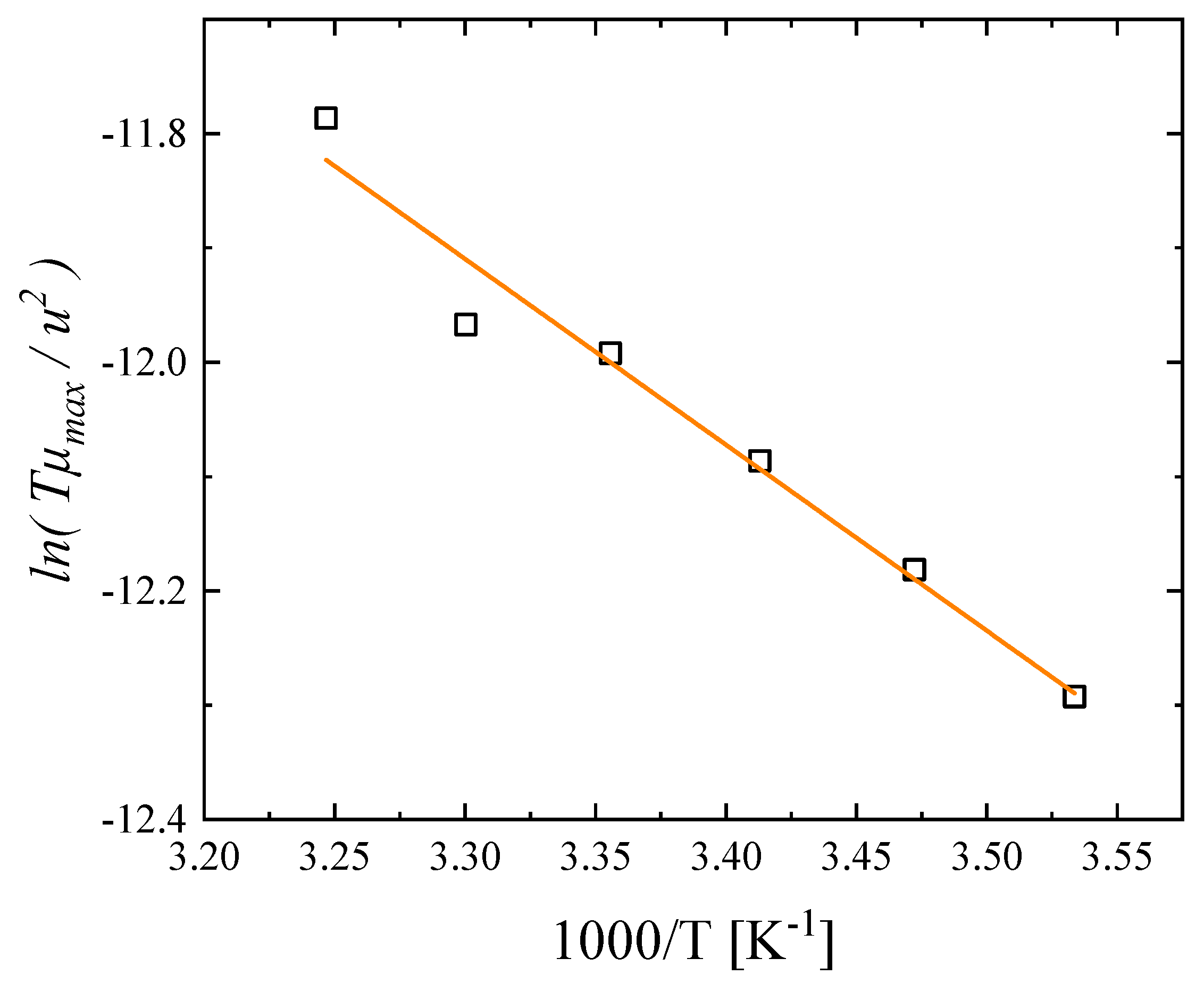
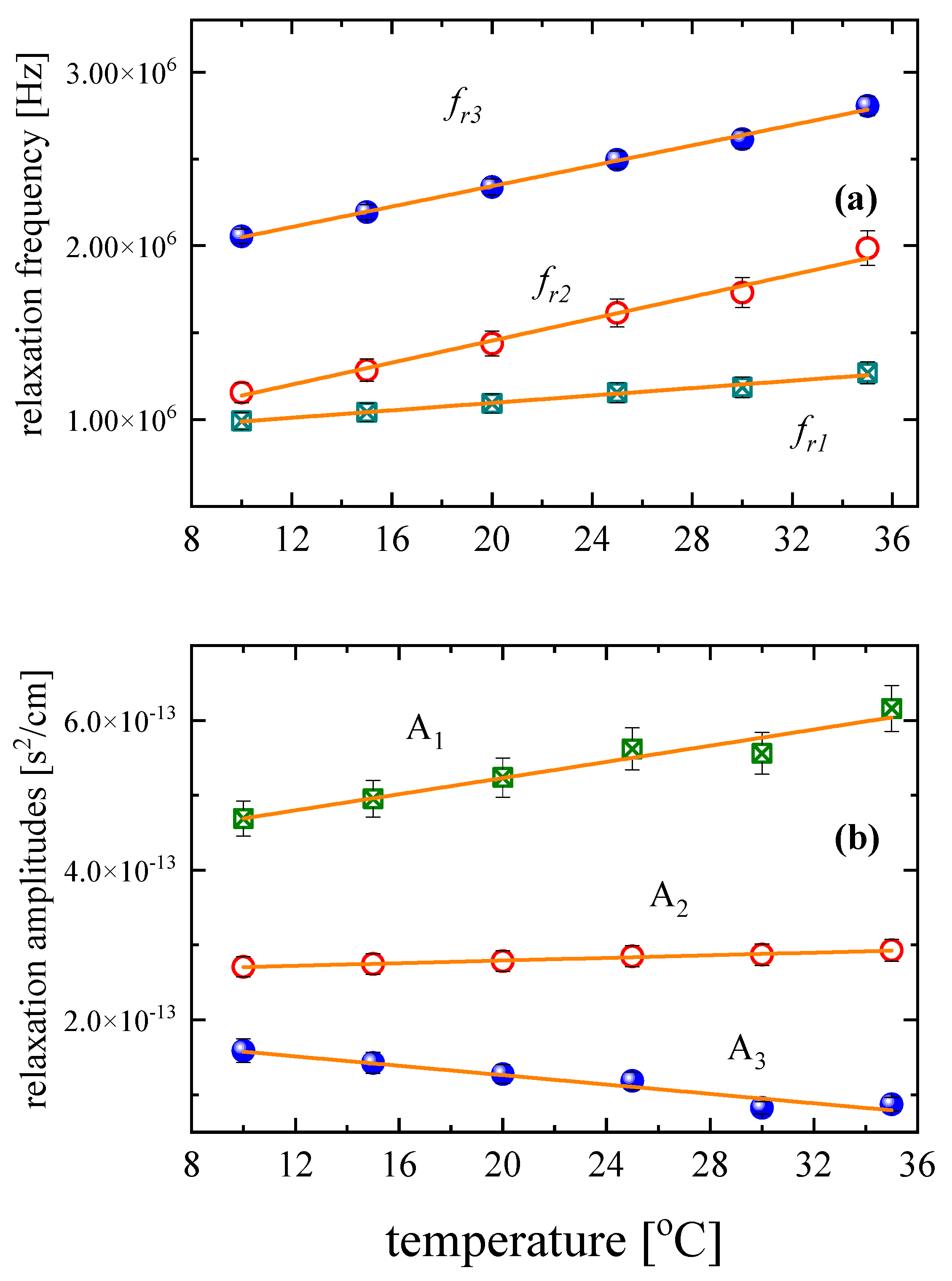
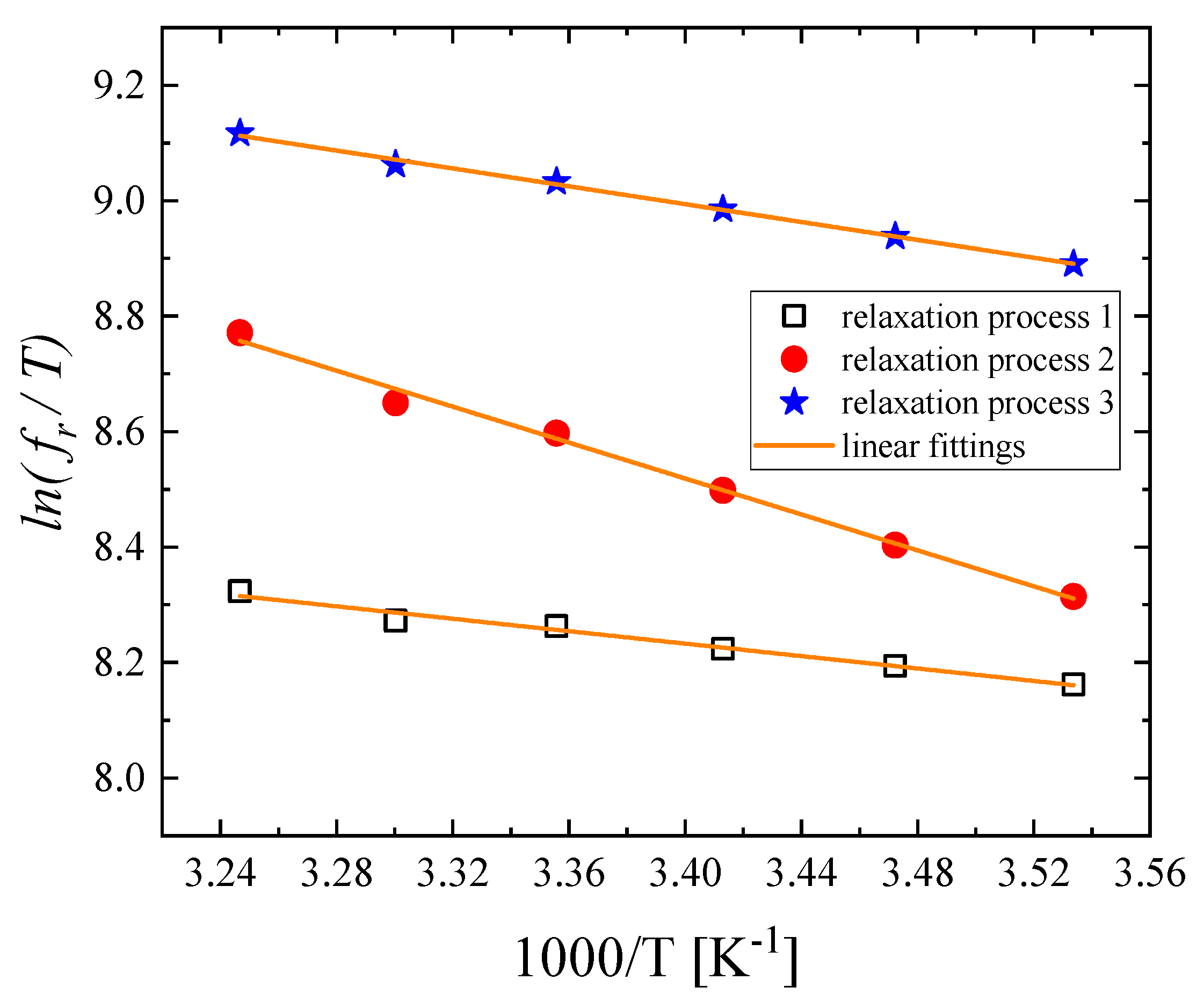
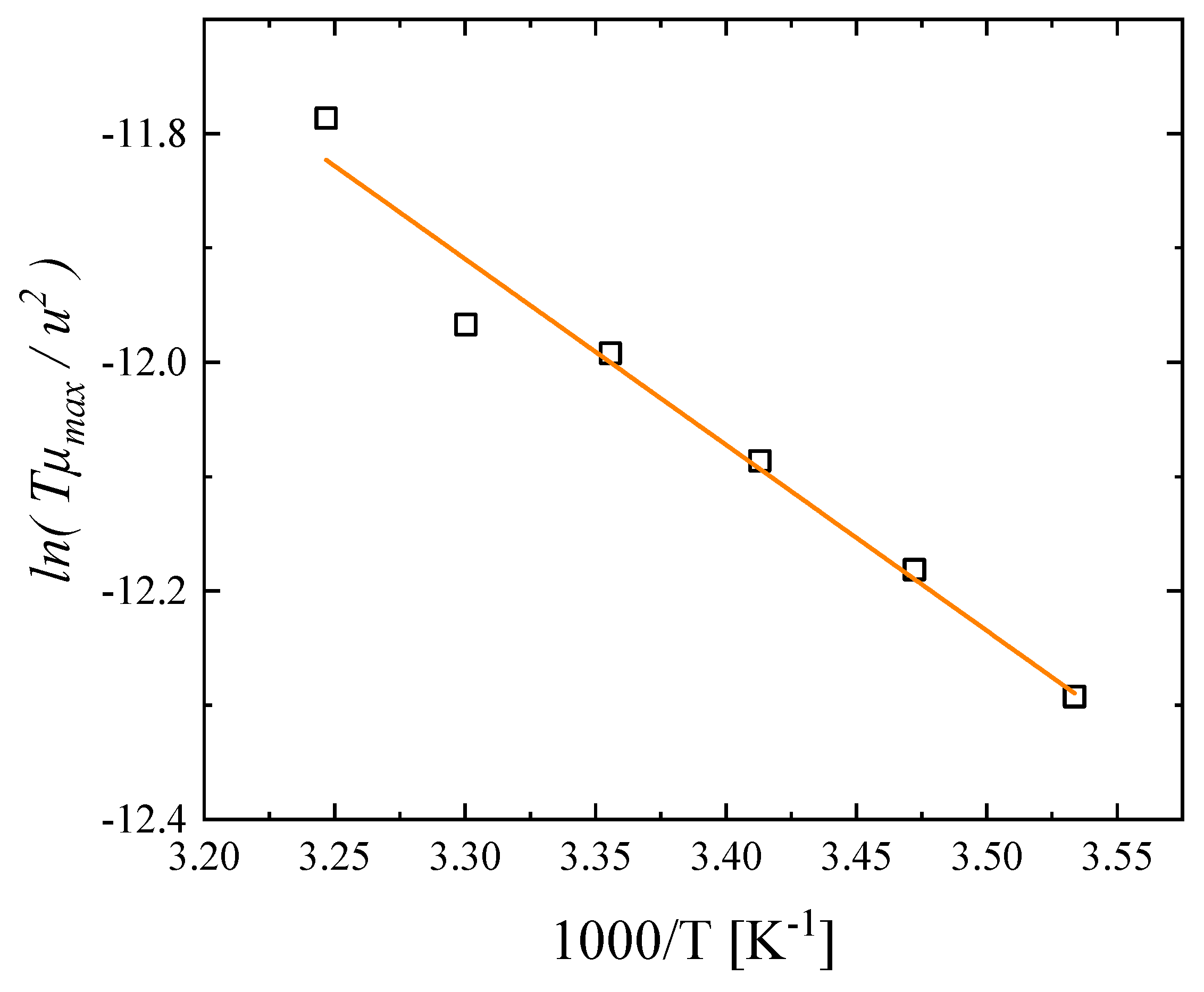
3.3. Effect of Temperature on the Relaxation Behavior
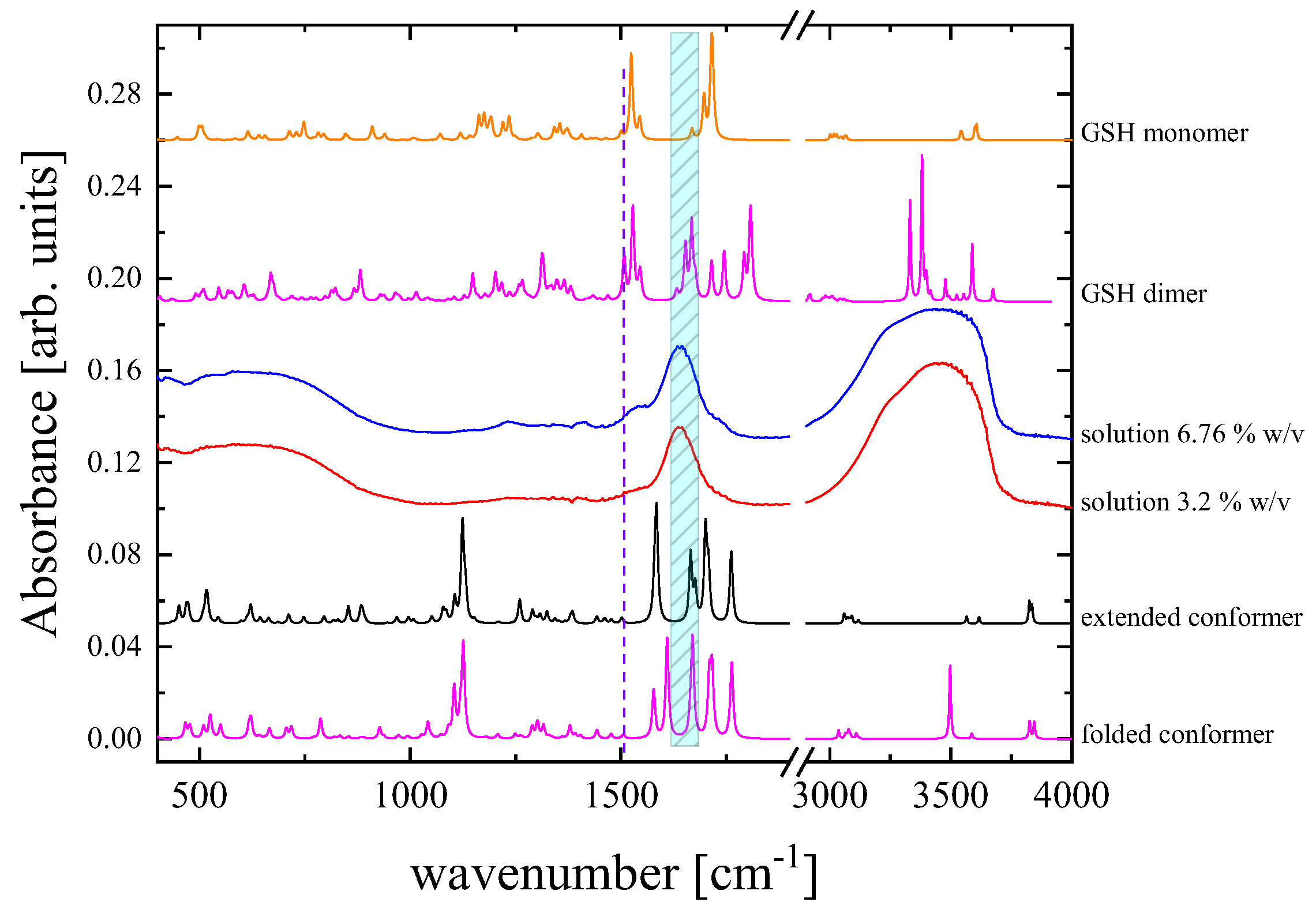
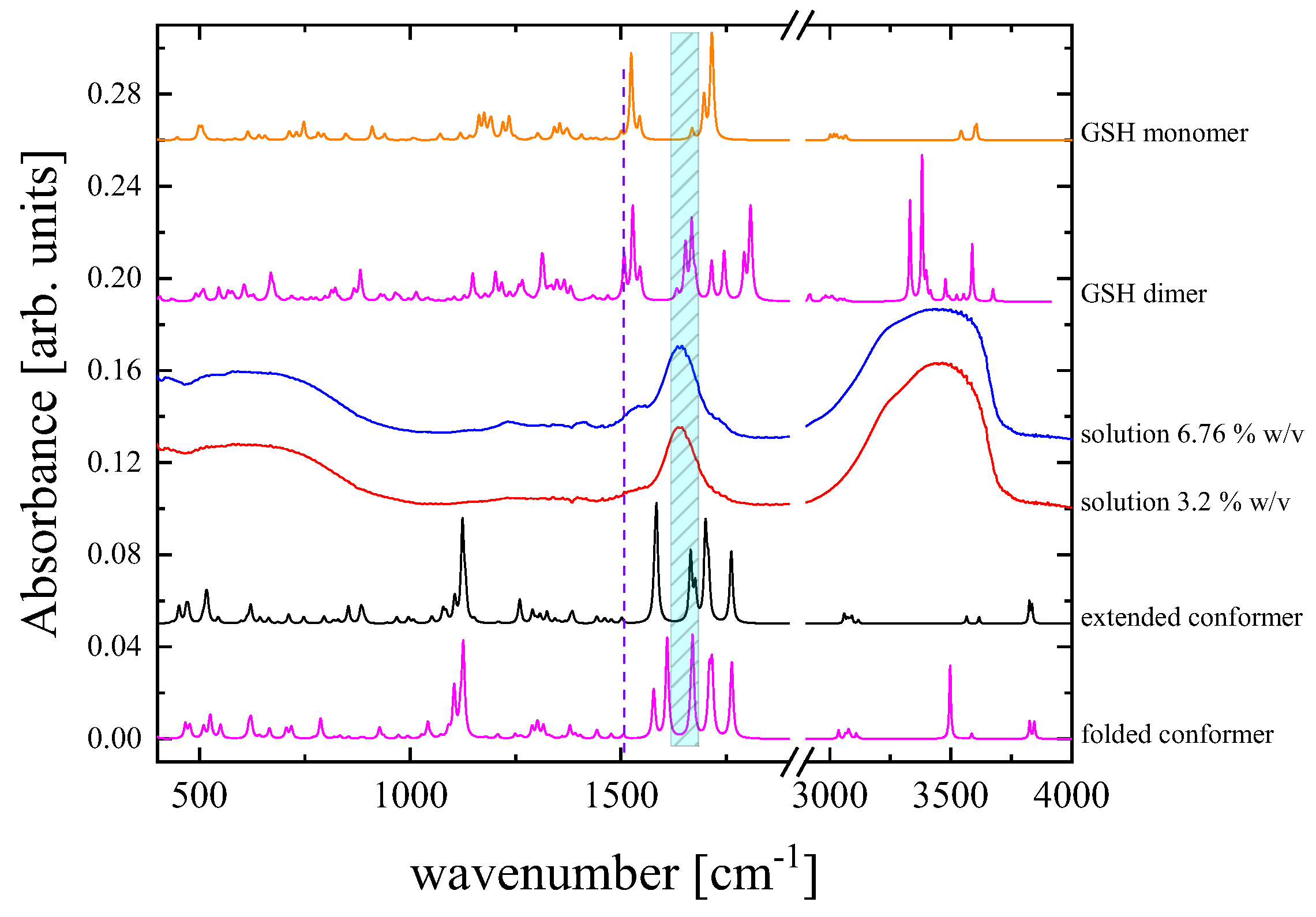
3.4. Probing Molecular Structure and Conformation based on Vibrational and Electronic Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunter, G.; Eagles, A. Glutathione: A critical study. J. Biol. Chem. 1927, 72, 147–166. [Google Scholar] [CrossRef]
- Hamilton, C.J.; Arbach, M.; Groom, M. Beyond Glutathione: Different Low Molecular Weight Thiols as Mediators of Redox Regulation and Other Metabolic Functions in Lower Organisms. In Recent Advances in Redox Active Plant and Microbial Products; Jacob, C., Kirsch, G., Slusarenko, A., Winyard, P., Burkholz, T., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 11, pp. 291–320. [Google Scholar]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef]
- Estrela, J.; Obrador, E.; Navarro, J.; Delavega, M.; Pellicer, J. Elimination of Ehrlich tumours by ATP-induced growth inhibition, glutathione depletion and X-rays. Nat. Med. 1995, 1, 84–88. [Google Scholar] [CrossRef]
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizário, J.E.; Menck, C.F.M. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef]
- Vargas, F.; Rodríguez-Gómez, I.; Pérez-Abud, R.; Vargas Tendero, P.; Baca, Y.; Wangensteen, R. Cardiovascular and renal manifestations of glutathione depletion induced by buthionine sulfoximine. Am. J. Hypertens. 2012, 25, 629–635. [Google Scholar] [CrossRef]
- Anderson, M.F.; Nilsson, M.; Eriksson, P.S.; Sims, N.R. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 2004, 354, 163–165. [Google Scholar] [CrossRef]
- Herzenberg, L.A.; De Rosa, S.C.; Dubs, J.G.; Roederer, M.; Anderson, M.T.; Ela, S.W.; Deresinski, S.C.; Herzenberg, L.A. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1967–1972. [Google Scholar] [CrossRef]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P.-T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef]
- Martin, H.L.; Teismann, P. Glutathione—A review on its role and significance in Parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2009, 1822, 625–630. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Jones, D.P.; Mody, V.C.; Carlson, J.L.; Lynn, M.J.; Sternberg, P. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002, 33, 1290–1300. [Google Scholar] [CrossRef]
- Witschi, A.; Reddy, S.; Stofer, B.; Lauterburg, B.H. The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 1992, 43, 667–669. [Google Scholar] [CrossRef]
- Siafarika, P.; Papanikolaou, M.G.; Kabanos, T.A.; Kalampounias, A.G. Probing the equilibrium between mono- and di-nuclear nickel(II)-diamidate {[NiII(DQPD)]x, x = 1, 2} complexes in chloroform solutions by combining acoustic and vibrational spectroscopies and molecular orbital calculations. Chem. Phys. 2021, 549, 111279. [Google Scholar] [CrossRef]
- Tsigoias, S.; Kouderis, C.; Mylona-Kosmas, A.; Boghosian, S.; Kalampounias, A.G. Proton-transfer in 1,1,3,3 tetramethyl guanidine by means of ultrasonic relaxation and Raman spectroscopies and molecular orbital calculations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 229, 117958. [Google Scholar] [CrossRef]
- Siafarika, P.; Kouderis, C.; Kalampounias, A.G. Non-Debye segmental relaxation of poly-N-vinyl-carbazole in dilute solution. Mol. Phys. 2021, 119, e1802075. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Kirillov, S.A.; Steffen, W.; Yannopoulos, S.N. Raman spectra and microscopic dynamics of bulk and confined salol. J. Mol. Struct. 2003, 651–653, 475–483. [Google Scholar] [CrossRef]
- Kalampounias, A.G. Dilution effect on the vibrational frequency and vibrational relaxation of PbCl2–KCl ionic liquids. J. Mol. Liq. 2015, 202, 68–74. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Tsilomelekis, G.; Boghosian, S. Unraveling the role of microenvironment and hydrodynamic forces on the vibrational relaxation rates of pyridine–water complexes. J. Mol. Liq. 2014, 198, 299–306. [Google Scholar] [CrossRef]
- Latsis, G.K.; Banti, C.N.; Kourkoumelis, N.; Papatriantafyllopoulou, C.; Panagiotou, N.; Tasiopoulos, A.; Douvalis, A.; Kalampounias, A.G.; Bakas, T.; Hadjikakou, S.K. Poly Organotin Acetates against DNA with Possible Implementation on Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 2055. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Papatheodorou, G.N. Ligand Field States and Vibrational Modes of Solid and Molten Elpasolite: Cs2NaHoCl6. Z. Naturforsch. 2007, 62, 169–175. [Google Scholar] [CrossRef]
- Stogiannidis, G.; Tsigoias, S.; Kalampounias, A.G. Conformational energy barriers in methyl acetate—Ethanol solutions: A temperature-dependent ultrasonic relaxation study and molecular orbital calculations. J. Mol. Liq. 2020, 302, 112519. [Google Scholar] [CrossRef]
- Kalampounias, A.G. Exploring conformational change profile of n-propyl ester of formic acid by combining ultrasonic relaxation spectroscopy and molecular orbital calculations. J. Mol. Struct. 2020, 1212, 128146. [Google Scholar] [CrossRef]
- Kouderis, C.; Siafarika, P.; Kalampounias, A.G. Disentangling proton-transfer and segmental motion relaxations in poly-vinyl-alcohol aqueous solutions by means of ultrasonic relaxation spectroscopy. Polymer 2021, 217, 123479. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kouderis, C.; Tsigoias, S.; Siafarika, P.; Kalampounias, A.G. The Effect of Alkali Iodide Salts in the Inclusion Process of Phenolphthalein in β-Cyclodextrin: A Spectroscopic and Theoretical Study. Molecules 2023, 28, 1147. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Gürsoy, O.; Smieško, M. Searching for bioactive conformations of drug-like ligands with current force fields: How good are we? J. Cheminform. 2017, 9, 29. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Singh, G.; Dogra, S.D.; Kaur, S.; Tripathi, S.K.; Prakash, S.; Rai, B.; Saini, G.S.S. Structure and vibrations of glutathione studied by vibrational spectroscopy and density functional theory. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 149, 505–515. [Google Scholar] [CrossRef]
- Fujiwara, S.; Formicka-Koziowska, G.; Kozlowski, H. Conformational Study of Glutathione by NMR. Bull. Chem. Soc. Jpn. 1977, 50, 3131–3135. [Google Scholar] [CrossRef]
- Lampela, O.; Juffer, A.H.; Rauk, A. Conformational Analysis of Glutathione in Aqueous Solution with Molecular Dynamics. J. Phys. Chem. A 2003, 107, 9208–9220. [Google Scholar] [CrossRef]
- Stevens, R.; Stevens, L.; Price, N.C. The stabilities of various thiol compounds used in protein purifications. Biochem. Educ. 1983, 11, 70. [Google Scholar] [CrossRef]
- Tryfon, A.; Siafarika, P.; Kouderis, C.; Kalampounias, A.G. Self-assembling of glutathione in aqueous environment: A combined experimental and theoretical study. J. Mol. Liq. 2023, 390, 122957. [Google Scholar] [CrossRef]
- Tsigoias, S.; Papanikolaou, M.G.; Kabanos, T.A.; Kalampounias, A.G. Structure and dynamics of aqueous norspermidine solutions: An in situ ultrasonic relaxation spectroscopic study. J. Phys. Condens. Matter 2021, 33, 495104. [Google Scholar] [CrossRef]
- Risva, M.; Tsigoias, S.; Boghosian, S.; Kaziannis, S.; Kalampounias, A.G. Exploring the influence of urea on the proton-transfer reaction in aqueous amine solutions with Raman and ultrasonic relaxation spectroscopy. Mol. Phys. 2023, 121, e2163314. [Google Scholar] [CrossRef]
- Nishikawa, S.; Yasunaga, T.; Takahashi, K. Kinetic studies of fast reactions in aqueous solutions of amylamine by means of ultrasonic absorption. Bull. Chem. Soc. Jpn. 1973, 46, 2992–2997. [Google Scholar] [CrossRef]
- Kouderis, C.; Siafarika, P.; Kalampounias, A.G. Molecular relaxation dynamics and self-association of dexamethasone sodium phosphate solutions. Chem. Pap. 2021, 75, 6115–6125. [Google Scholar] [CrossRef]
- Kaatze, U.; Hushcha, T.O.; Eggers, F. Ultrasonic broadband spectrometry of liquids a research tool in pure and applied chemistry and chemical physics. J. Solut. Chem. 2000, 29, 299–368. [Google Scholar] [CrossRef]
- Herzfeld, K.F.; Litovitz, T.A. Absorption and Dispersion of Ultrasonic Waves, 1st ed.; Academic Press: New York, NY, USA, 1959. [Google Scholar]
- Ensminger, D.; Bond, L.J. Ultrasonics: Fundamentals, Technologies, and Applications, 3rd ed.; CRC Press: New York, NY, USA, 2011. [Google Scholar]
- Blandamer, M.J. Introduction to Chemical Ultrasonics; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Nishikawa, S.; Haraguchi, H.; Fukuyama, Y. Effect of ether oxygen on proton transfer and aggregation reactions of amines in water by ultrasonic absorption method. Bull. Chem. Soc. Jpn. 1991, 64, 1274–1282. [Google Scholar] [CrossRef]
- Nishikawa, S.; Kamimura, E. Dynamic characteristic of amitriptyline in water by ultrasonic relaxation method and molecular orbital calculation. J. Phys. Chem. A 2011, 115, 535–539. [Google Scholar] [CrossRef]
- Beaven, G.H.; Holiday, E.R. Ultraviolet Absorption Spectra of Proteins and Amino Acids. Adv. Protein Chem. 1952, 7, 319–386. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| c (mM) | A1 (×10−13 s2/cm) | fr1 (MHz) | A2 (×10−13 s2/cm) | fr2 (MHz) | A3 (×10−13 s2/cm) | fr3 (MHz) | Β (×10−16 s2/cm) |
|---|---|---|---|---|---|---|---|
| 1 | 4.00 | 1.170 | 2.07 | 1.937 | 1.37 | 2.468 | 2.50 |
| 2 | 4.51 | 1.071 | 2.41 | 1.694 | 1.30 | 2.465 | 2.50 |
| 3 | 4.47 | 1.077 | 2.49 | 1.654 | 1.30 | 2.475 | 2.50 |
| 4 | 4.98 | 1.071 | 2.50 | 1.565 | 1.28 | 2.347 | 2.50 |
| 5 | 5.24 | 1.092 | 2.78 | 1.438 | 1.28 | 2.372 | 2.50 |
| 7.5 | 5.95 | 1.050 | 3.31 | 1.258 | 1.23 | 2.317 | 2.50 |
| 10 | 6.83 | 1.075 | 3.97 | 1.044 | 1.19 | 1.998 | 2.50 |
| Τ (°C) | A1 (×10−13 s2/cm) | fr1 (MHz) | A2 (×10−13 s2/cm) | fr2 (MHz) | A3 (×10−13 s2/cm) | fr3 (MHz) | Β (×10−16 s2/cm) |
|---|---|---|---|---|---|---|---|
| 10 | 4.69 | 0.992 | 2.71 | 1.155 | 1.59 | 2.055 | 2.50 |
| 15 | 4.95 | 1.042 | 2.75 | 1.284 | 1.43 | 2.194 | 2.50 |
| 20 | 5.24 | 1.092 | 2.78 | 1.438 | 1.28 | 2.372 | 2.50 |
| 25 | 5.62 | 1.155 | 2.85 | 1.614 | 1.18 | 2.495 | 2.50 |
| 30 | 5.56 | 1.186 | 2.87 | 1.730 | 8.25 | 2.613 | 2.50 |
| 35 | 6.16 | 1.269 | 2.93 | 1.986 | 8.73 | 2.805 | 2.50 |
| Absorption Band Frequency (cm−1) | Assignment to Specific Functional Groups |
|---|---|
| 1598 | –C=O amide |
| 1712 | –C=O acid |
| 2523 | –SH |
| 3020 | C–H |
| 3250–3628 | –OH, NH, NH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tryfon, A.; Siafarika, P.; Kouderis, C.; Kalampounias, A.G. Insight into the Structural and Dynamical Processes of Peptides by Means of Vibrational and Ultrasonic Relaxation Spectroscopies, Molecular Docking, and Density Functional Theory Calculations. ChemEngineering 2024, 8, 21. https://doi.org/10.3390/chemengineering8010021
Tryfon A, Siafarika P, Kouderis C, Kalampounias AG. Insight into the Structural and Dynamical Processes of Peptides by Means of Vibrational and Ultrasonic Relaxation Spectroscopies, Molecular Docking, and Density Functional Theory Calculations. ChemEngineering. 2024; 8(1):21. https://doi.org/10.3390/chemengineering8010021
Chicago/Turabian StyleTryfon, Afrodite, Panagiota Siafarika, Constantine Kouderis, and Angelos G. Kalampounias. 2024. "Insight into the Structural and Dynamical Processes of Peptides by Means of Vibrational and Ultrasonic Relaxation Spectroscopies, Molecular Docking, and Density Functional Theory Calculations" ChemEngineering 8, no. 1: 21. https://doi.org/10.3390/chemengineering8010021
APA StyleTryfon, A., Siafarika, P., Kouderis, C., & Kalampounias, A. G. (2024). Insight into the Structural and Dynamical Processes of Peptides by Means of Vibrational and Ultrasonic Relaxation Spectroscopies, Molecular Docking, and Density Functional Theory Calculations. ChemEngineering, 8(1), 21. https://doi.org/10.3390/chemengineering8010021