
Two-Step Conversion of CO2 to Light Olefins: Laboratory-Scale Demonstration and Scale-Up Considerations

 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Experimental Setup
2.3. Experimental Work
2.4. Calculation Methods
3. Results
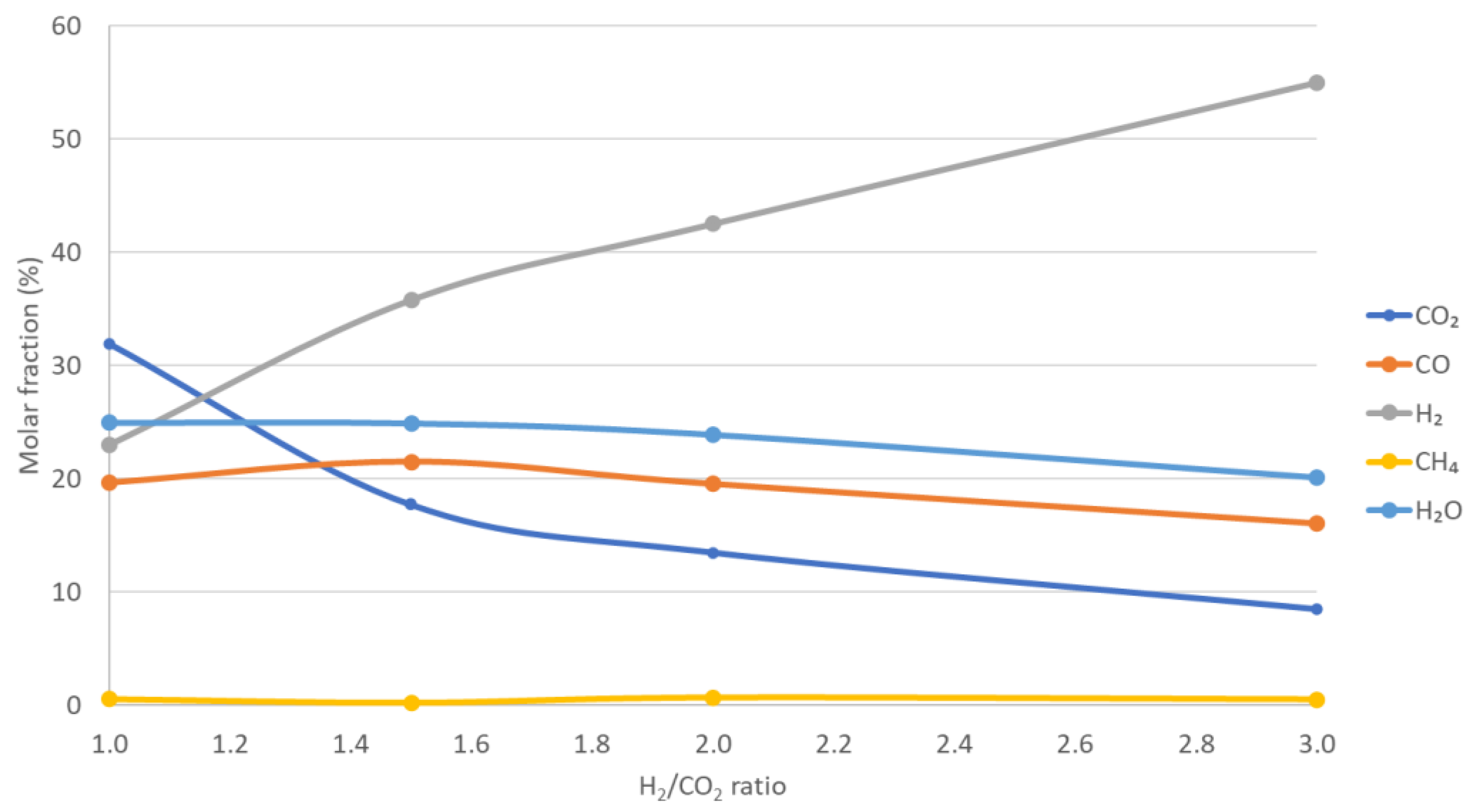
3.1. Reverse Water Gas Shift Results
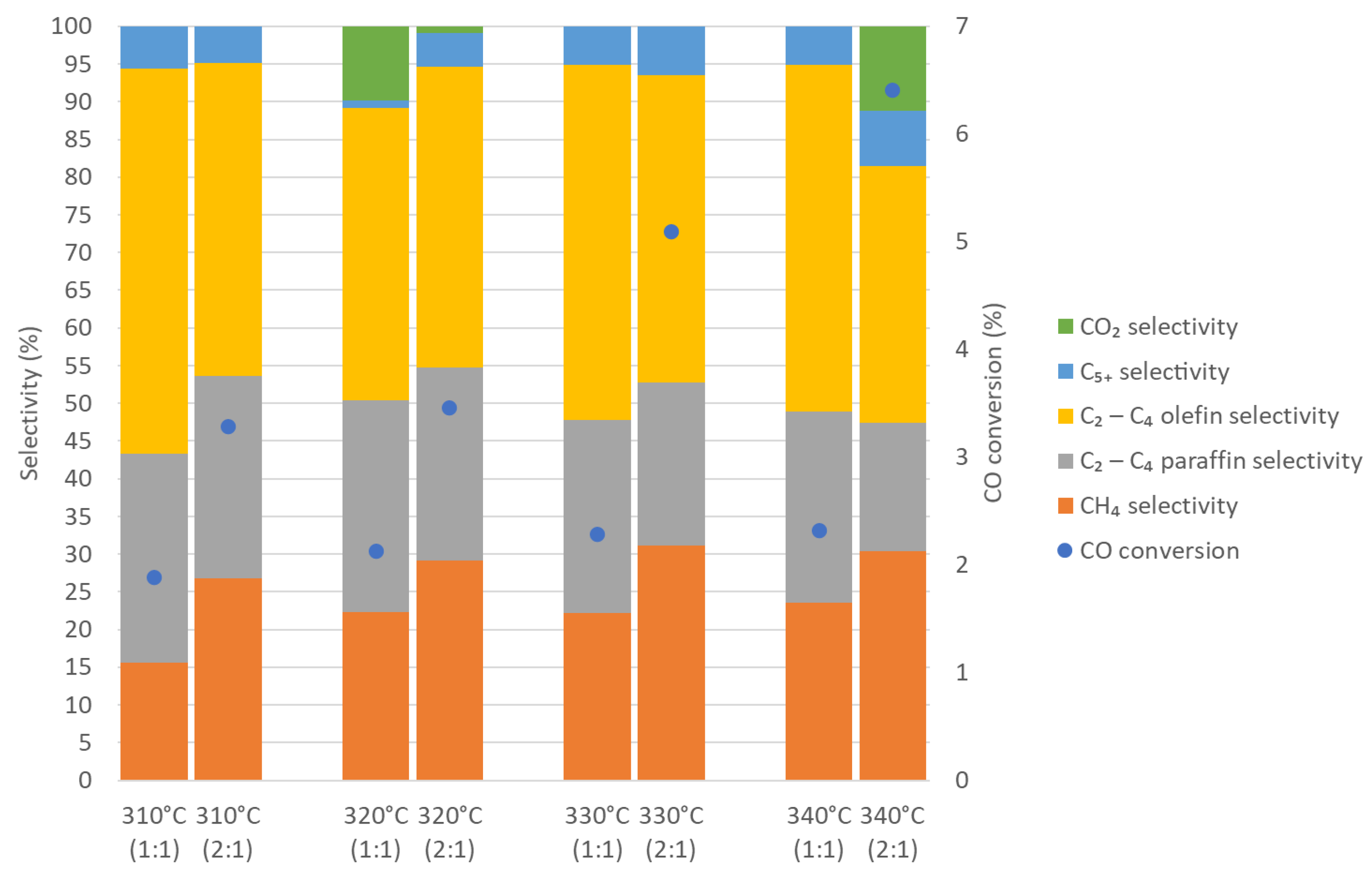
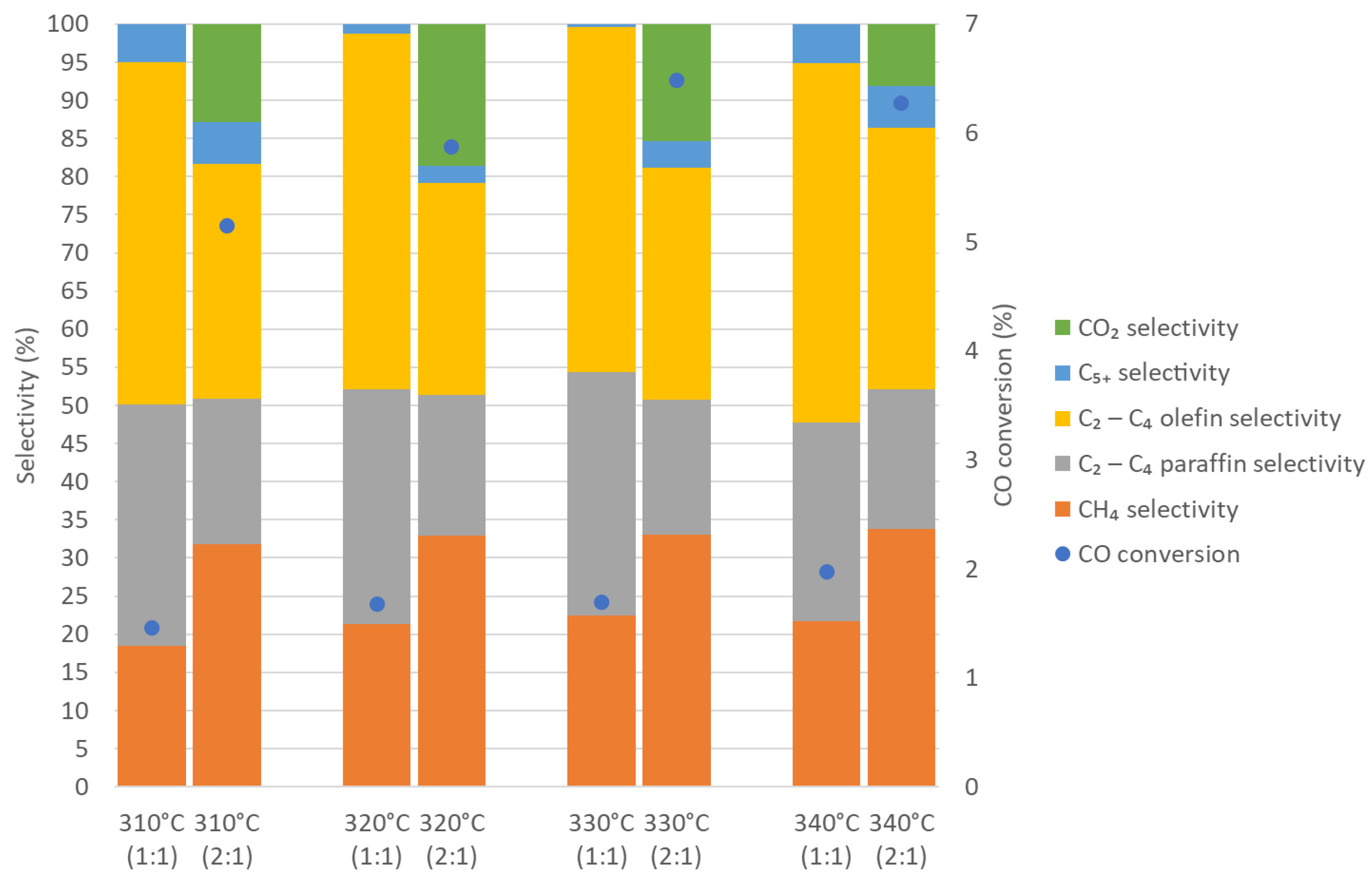
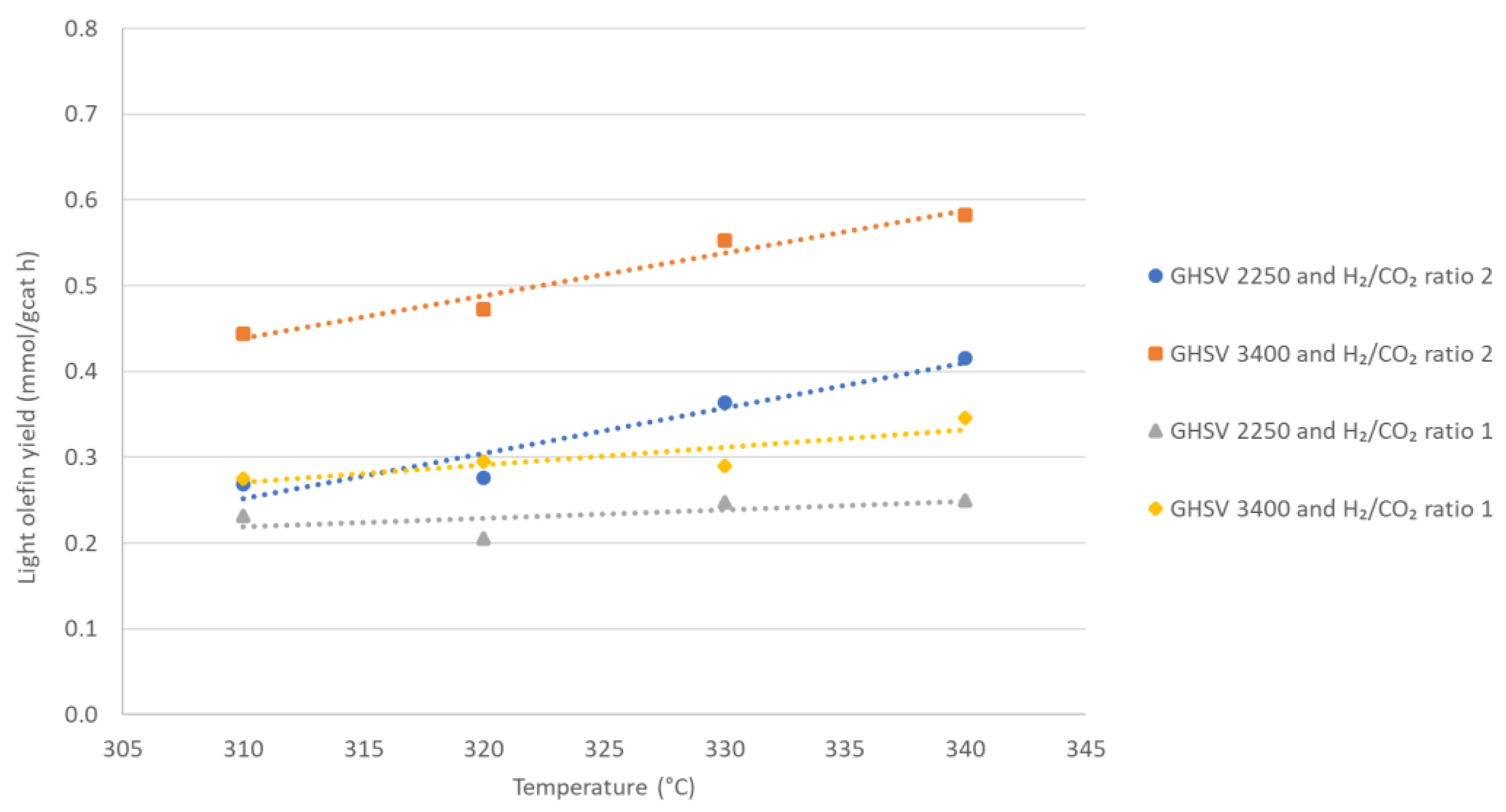
3.2. Fischer–Tropsch Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Akah, A.; Williams, J.; Ghrami, M. An Overview of Light Olefins Production via Steam Enhanced Catalytic Cracking. Catal. Surv. Asia 2019, 23, 265–276. [Google Scholar] [CrossRef]
- Corma, A.; Corresa, E.; Mathieu, Y.; Sauvanaud, L.; Al-Bogami, S.; Al-Ghrami, M.S.; Bourane, A. Crude Oil to Chemicals: Light Olefins from Crude Oil. Catal. Sci. Technol. 2017, 7, 12–46. [Google Scholar] [CrossRef] [Green Version]
- Amghizar, I.; Vandewalle, L.A.; van Geem, K.M.; Marin, G.B. New Trends in Olefin Production. Engineering 2017, 3, 171–178. [Google Scholar] [CrossRef]
- Zimmermann, H. Light Olefins—Challenges from New Production Routes? In Opportunities and Challenges at the Interface between Petrochemistry and Refinery; U.S. Department of Energy: Washington, DC, USA, 2007; pp. 75–86. [Google Scholar]
- Numpilai, T.; Cheng, C.K.; Limtrakul, J.; Witoon, T. Recent Advances in Light Olefins Production from Catalytic Hydrogenation of Carbon Dioxide. Process Saf. Environ. Prot. 2021, 151, 401–427. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef] [Green Version]
- Marlin, D.S.; Sarron, E.; Sigurbjörnsson, Ó. Process Advantages of Direct CO2 to Methanol Synthesis. Front. Chem. 2018, 6, 446. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on Methanation—From Fundamentals to Current Projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Fazlollahi, F.; Nikoo, M.K.; Sefidkon, F.; Klemeš, J.J.; Baxter, L.L. Mechanisms and Kinetics of CO2 Hydrogenation to Value-Added Products: A Detailed Review on Current Status and Future Trends. Renew. Sustain. Energy Rev. 2017, 80, 1292–1311. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Yang, X.; Zhao, B.; Huang, Y. Designing of Highly Selective and High-Temperature Endurable RWGS Heterogeneous Catalysts: Recent Advances and the Future Directions. J. Energy Chem. 2017, 26, 854–867. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.W.; Shi, C.; Ma, D. Highly Dispersed Copper over β-Mo2C as an Efficient and Stable Catalyst for the Reverse Water Gas Shift (RWGS) Reaction. ACS Catal. 2017, 7, 912–918. [Google Scholar] [CrossRef]
- Zhou, G.; Xie, F.; Deng, L.; Zhang, G.; Xie, H. Supported Mesoporous Cu/CeO2-δ Catalyst for CO2 Reverse Water–Gas Shift Reaction to Syngas. Int. J. Hydrogen Energy 2020, 45, 11380–11393. [Google Scholar] [CrossRef]
- Zhao, K.; Bkour, Q.; Hou, X.; Kang, S.W.; Park, J.C.; Norton, M.G.; Yang, J.-I.; Ha, S. Reverse Water Gas Shift Reaction over CuFe/Al2O3 Catalyst in Solid Oxide Electrolysis Cell. Chem. Eng. J. 2018, 336, 20–27. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Héroguel, F.; Klllç, M.; Baranowski, C.J.; Artiglia, L.; Röthlisberger, U.; Luterbacher, J.S.; Kröcher, O. Cu-Al Spinel as a Highly Active and Stable Catalyst for the Reverse Water Gas Shift Reaction. ACS Catal. 2019, 9, 6243–6251. [Google Scholar] [CrossRef]
- Benzinger, W.; Daymo, E.; Hettel, M.; Maier, L.; Antinori, C.; Pfeifer, P.; Deutschmann, O. Reverse Water Gas Shift (RWGS) over Ni—Spatially-Resolved Measurements and Simulations. Chem. Eng. J. 2019, 362, 430–441. [Google Scholar] [CrossRef]
- Wolf, A.; Jess, A.; Kern, C. Syngas Production via Reverse Water-Gas Shift Reaction over a Ni-Al2O3 Catalyst: Catalyst Stability, Reaction Kinetics, and Modeling. Chem. Eng. Technol. 2016, 39, 1040–1048. [Google Scholar] [CrossRef]
- Li, S.; Xu, Y.; Chen, Y.; Li, W.; Lin, L.; Li, M.; Deng, Y.; Wang, X.; Ge, B.; Yang, C.; et al. Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal–Support Interaction. Angew. Chem. Int. Ed. 2017, 56, 10761–10765. [Google Scholar] [CrossRef]
- Kaiser, P.; Unde, R.B.; Kern, C.; Jess, A. Production of Liquid Hydrocarbons with CO2 as Carbon Source Based on Reverse Water-Gas Shift and Fischer-Tropsch Synthesis. Chem. Ing. Tech. 2013, 85, 489–499. [Google Scholar] [CrossRef]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. Highly Efficient Ni/CeO2-Al2O3 Catalysts for CO2 Upgrading via Reverse Water-Gas Shift: Effect of Selected Transition Metal Promoters. Appl. Catal. B Environ. 2018, 232, 464–471. [Google Scholar] [CrossRef]
- Dzuryk, S.; Rezaei, E. Intensification of the Reverse Water Gas Shift Reaction by Water-Permeable Packed-Bed Membrane Reactors. Ind. Eng. Chem. Res. 2020, 59, 18907–18920. [Google Scholar] [CrossRef]
- Samimi, F.; Hamedi, N.; Rahimpour, M.R. Green Methanol Production Process from Indirect CO2 Conversion: RWGS Reactor versus RWGS Membrane Reactor. J. Environ. Chem. Eng. 2019, 7, 102813. [Google Scholar] [CrossRef]
- Ghodhbene, M.; Bougie, F.; Fongarland, P.; Iliuta, M.C. Hydrophilic Zeolite Sorbents for In-Situ Water Removal in High Temperature Processes. Can. J. Chem. Eng. 2017, 95, 1842–1849. [Google Scholar] [CrossRef]
- Lee, M.; Kim, Y.; Lim, H.S.; Jo, A.; Kang, D.; Lee, J.W. Reverse Water–Gas Shift Chemical Looping Using a Core–Shell Structured Perovskite Oxygen Carrier. Energies 2020, 13, 5324. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kent, R.A.; Yung, M.M.; Kuhn, J.N. Carbon Dioxide Conversion by Reverse Water-Gas Shift Chemical Looping on Perovskite-Type Oxides. Ind. Eng. Chem. Res. 2014, 53, 5828–5837. [Google Scholar] [CrossRef]
- Wenzel, M.; Rihko-Struckmann, L.; Sundmacher, K. Continuous Production of CO from CO2 by RWGS Chemical Looping in Fixed and Fluidized Bed Reactors. Chem. Eng. J. 2018, 336, 278–296. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sun, Y.; Wang, B.; Xiao, H.; Wu, J.; Chen, L.; Fu, M.; Ye, D. Effect of Plasma on Catalytic Conversion of CO2 with Hydrogen over Pd/ZnO in a Dielectric Barrier Discharge Reactor. J. Phys. D Appl. Phys. 2019, 52, 244001. [Google Scholar] [CrossRef]
- Zeng, Y.; Tu, X. Plasma-Catalytic CO2 Hydrogenation at Low Temperatures. IEEE Trans. Plasma Sci. 2016, 44, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Oshima, K.; Shinagawa, T.; Nogami, Y.; Manabe, R.; Ogo, S.; Sekine, Y. Low Temperature Catalytic Reverse Water Gas Shift Reaction Assisted by an Electric Field. Catal. Today 2014, 232, 27–32. [Google Scholar] [CrossRef]
- Klerk, A.D. Fischer-Tropsch Refining; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2011. [Google Scholar]
- Maitlis, P.M.; de Klerk, A. Greener Fischer-Tropsch Processes; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2012. [Google Scholar]
- Shafer, W.D.; Gnanamani, M.K.; Graham, U.M.; Yang, J.; Masuku, C.M.; Jacobs, G.; Davis, B.H. Fischer-Tropsch: Product Selectivity-the Fingerprint of Synthetic Fuels. Catalysts 2019, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.A.; Nwaoha, C.; Towler, B.F. Gas-to-Liquids (GTL): A Review of an Industry Offering Several Routes for Monetizing Natural Gas. J. Nat. Gas Sci. Eng. 2012, 9, 196–208. [Google Scholar] [CrossRef]
- Wilhelm, D.J.; Simbeck, D.R.; Karp, A.D.; Dickenson, R.L. Syngas Production for Gas-to-Liquids Applications: Technologies, Issues and Outlook. Fuel Process. Technol. 2001, 71, 139–148. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, S.; Li, Y. Coal Liquefaction Technologies-Development in China and Challenges in Chemical Reaction Engineering. Chem. Eng. Sci. 2010, 65, 12–17. [Google Scholar] [CrossRef]
- Rafati, M.; Wang, L.; Dayton, D.C.; Schimmel, K.; Kabadi, V.; Shahbazi, A. Techno-Economic Analysis of Production of Fischer-Tropsch Liquids via Biomass Gasification: The Effects of Fischer-Tropsch Catalysts and Natural Gas Co-Feeding. Energy Convers. Manag. 2017, 133, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.M.; Platon, A.; Satrio, J.A.; Brown, R.C. Techno-Economic Analysis of Biomass-to-Liquids Production Based on Gasification. Fuel 2010, 89 (Suppl. 1), S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Ngamcharussrivichai, C.; Liu, X.; Li, X.; Vitidsant, T.; Fujimoto, K. An Active and Selective Production of Gasoline-Range Hydrocarbons over Bifunctional Co-Based Catalysts. Fuel 2007, 86, 50–59. [Google Scholar] [CrossRef]
- Botes, G.F.; Bromfield, T.C.; Coetzer, R.L.J.; Crous, R.; Gibson, P.; Ferreira, A.C. Development of a Chemical Selective Iron Fischer Tropsch Catalyst. Catal. Today 2016, 275, 40–48. [Google Scholar] [CrossRef]
- Sun, J.; Yang, G.; Peng, X.; Kang, J.; Wu, J.; Liu, G.; Tsubaki, N. Beyond Cars: Fischer-Tropsch Synthesis for Non-Automotive Applications. ChemCatChem 2019, 11, 1412–1424. [Google Scholar] [CrossRef]
- Todic, B.; Nowicki, L.; Nikacevic, N.; Bukur, D.B. Fischer-Tropsch Synthesis Product Selectivity over an Industrial Iron-Based Catalyst: Effect of Process Conditions. Catal. Today 2016, 261, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Gong, K.; Wang, C.; An, Y.; Wang, X.; Qi, X.; Li, S.; Lu, Y.; Zhong, L.; Sun, Y. Fischer-Tropsch Synthesis to Olefins: Catalytic Performance and Structure Evolution of Co2C-Based Catalysts under a CO2 Environment. ACS Catal. 2019, 9, 9554–9567. [Google Scholar] [CrossRef]
- Xie, J.; Yang, J.; Dugulan, A.I.; Holmen, A.; Chen, D.; de Jong, K.P.; Louwerse, M.J. Size and Promoter Effects in Supported Iron Fischer-Tropsch Catalysts: Insights from Experiment and Theory. ACS Catal. 2016, 6, 3147–3157. [Google Scholar] [CrossRef]
- Han, Z.; Qian, W.; Zhang, H.; Ma, H.; Sun, Q.; Ying, W. Effect of Rare-Earth Promoters on Precipitated Iron-Based Catalysts for Fischer-Tropsch Synthesis. Ind. Eng. Chem. Res. 2020, 59, 14598–14605. [Google Scholar] [CrossRef]
- Wu, X.; Ma, H.; Zhang, H.; Qian, W.; Liu, D.; Sun, Q.; Ying, W. High-Temperature Fischer-Tropsch Synthesis of Light Olefins over Nano-Fe3O4@MnO2 Core-Shell Catalysts. Ind. Eng. Chem. Res. 2019, 58, 21350–21362. [Google Scholar] [CrossRef]
- Portillo, A.; Ateka, A.; Ereña, J.; Aguayo, A.T.; Bilbao, J. Conditions for the Joint Conversion of CO2 and Syngas in the Direct Synthesis of Light Olefins Using In2O3-ZrO2/SAPO-34 Catalyst. Ind. Eng. Chem. Res. 2021, 61, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jia, G.; Zhao, C.; Xing, Y. Selective Iron Catalysts for Direct Fischer-Tropsch Synthesis to Light Olefins. Ind. Eng. Chem. Res. 2021, 60, 6137–6146. [Google Scholar] [CrossRef]
- Dewilde, J.F.; Ho, C.R.; Conner, J.; Smith, A.; Kirilin, A.V.; Malek, A.; Witt, P.M. Kinetics of Direct Olefin Synthesis from Syngas over Mixed Beds of Zn-Zr Oxides and SAPO-34. Ind. Eng. Chem. Res. 2021, 60, 14166–14175. [Google Scholar] [CrossRef]
- Zhang, P.; Meng, F.; Yang, L.; Yang, G.; Liang, X.; Li, Z. Syngas to Olefins over a CrMnGa/SAPO-34 Bifunctional Catalyst: Effect of Cr and Cr/Mn Ratio. Ind. Eng. Chem. Res. 2021, 60, 13214–13222. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; Koeken, A.C.J.; Bitter, J.H.; Davidian, T.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Effect of Precursor on the Catalytic Performance of Supported Iron Catalysts for the Fischer-Tropsch Synthesis of Lower Olefins. Catal. Today 2013, 215, 95–102. [Google Scholar] [CrossRef]
- Bukur, D.B.; Todic, B.; Elbashir, N. Role of Water-Gas-Shift Reaction in Fischer–Tropsch Synthesis on Iron Catalysts: A Review. Catal. Today 2016, 275, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Hamdeh, H.; Davis, B.H. Fischer-Tropsch Synthesis: Catalyst activation of low alpha iron catalyst. Catal. Today 2009, 140, 127–134. [Google Scholar] [CrossRef]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A Study on the Effect of Support’s Reducibility on the Reverse Water-Gas Shift Reaction over Pt Catalysts. Appl. Catal. A Gen. 2012, 423–424, 100–107. [Google Scholar] [CrossRef]
- Kaisalo, N.; Simell, P.; Frilund, C.; Vidal Vazquez, F.; Hannula, I. Method and Apparatus for Producing Carbon Monoxide. U.S. Patent US20210246034A1, 12 March 2019. [Google Scholar]
- Gong, W.; Ye, R.P.; Ding, J.; Wang, T.; Shi, X.; Russell, C.K.; Tang, J.; Eddings, E.G.; Zhang, Y.; Fan, M. Effect of Copper on Highly Effective Fe-Mn Based Catalysts during Production of Light Olefins via Fischer-Tropsch Process with Low CO2 Emission. Appl. Catal. B Environ. 2020, 278, 119302. [Google Scholar] [CrossRef]
- Song, F.; Yong, X.; Wu, X.; Zhang, W.; Ma, Q.; Zhao, T.; Tan, M.; Guo, Z.; Zhao, H.; Yang, G.; et al. FeMn@HZSM-5 Capsule Catalyst for Light Olefins Direct Synthesis via Fischer-Tropsch Synthesis: Studies on Depressing the CO2 Formation. Appl. Catal. B Environ. 2022, 300, 120713. [Google Scholar] [CrossRef]
- Liu, Y.; Teng, B.T.; Guo, X.H.; Li, Y.; Chang, J.; Tian, L.; Hao, X.; Wang, Y.; Xiang, H.W.; Xu, Y.Y.; et al. Effect of Reaction Conditions on the Catalytic Performance of Fe-Mn Catalyst for Fischer-Tropsch Synthesis. J. Mol. Catal. A Chem. 2007, 272, 182–190. [Google Scholar] [CrossRef]
- Yahyazadeh, A.; Dalai, A.K.; Ma, W.; Zhang, L. Fischer–Tropsch Synthesis for Light Olefins from Syngas: A Review of Catalyst Development. Reactions 2021, 2, 227–257. [Google Scholar] [CrossRef]
- Turan, A.Z.; Ataç, Ö.; Kurucu, O.A.; Ersöz, A.; Sarıoğlan, A.; Okutan, H. Kinetic Modeling of Fischer–Tropsch-to-Olefins Process via Advanced Optimization. Int. J. Chem. Kinet. 2022, 54, 3–15. [Google Scholar] [CrossRef]
- Marchese, M.; Giglio, E.; Santarelli, M.; Lanzini, A. Energy Performance of Power-to-Liquid Applications Integrating Biogas Upgrading, Reverse Water Gas Shift, Solid Oxide Electrolysis and Fischer-Tropsch Technologies. Energy Convers. Manag. X 2020, 6, 100041. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | RWGS Reactor | FT Reactor |
|---|---|---|
| Reactor length (mm) | 255 | 710 |
| Reactor inside diameter (mm) | 4 | 6 |
| Temperature (°C) | 850 | 310–340 |
| GHSV (h−1) | 20,000–50,000 | 2000–3500 |
| Pressure/bar | 6 | |
| H2/CO2 and H2/CO-molar ratio | 1–3 | |
| RWGS Inlet | RWGS Outlet, FT Inlet | ||
|---|---|---|---|
| Volumetric Flow Rate (NL/min) | H2/CO2 Ratio | Volumetric Flow Rate (NL/min) | H2/CO Ratio |
| 1 | 2.0 | 0.7 | 2.0 |
| 1 | 1.5 | 0.8 | 1.5 |
| 1 | 1.0 | 1 | 1.1 |
| 1.5 | 2.0 | 1.1 | 2.2 |
| 1.5 | 1.5 | 1.3 | 1.7 |
| 1.5 | 1.0 | 1.4 | 1.2 |
| 2 | 2.0 | 1.5 | 2.4 |
| 2 | 1.5 | 1.7 | 1.9 |
| 2 | 1.0 | 1.9 | 1.4 |
| GHSV (h−1) | 2250 | 2250 | 3400 | 3400 |
|---|---|---|---|---|
| H2/CO2 molar ratio | 1 | 2 | 1 | 2 |
| CO conversion (%) | 2.1 | 3.5 | 1.7 | 5.9 |
| CH4 selectivity (%) | 22.4 | 29.1 | 21.3 | 32.9 |
| Light olefin selectivity (%) | 39.9 | 39.8 | 46.5 | 27.8 |
| Light paraffin selectivity (%) | 28.0 | 25.7 | 30.9 | 18.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinikainen, M.; Braunschweiler, A.; Korpilo, S.; Simell, P.; Alopaeus, V. Two-Step Conversion of CO2 to Light Olefins: Laboratory-Scale Demonstration and Scale-Up Considerations. ChemEngineering 2022, 6, 96. https://doi.org/10.3390/chemengineering6060096
Reinikainen M, Braunschweiler A, Korpilo S, Simell P, Alopaeus V. Two-Step Conversion of CO2 to Light Olefins: Laboratory-Scale Demonstration and Scale-Up Considerations. ChemEngineering. 2022; 6(6):96. https://doi.org/10.3390/chemengineering6060096
Chicago/Turabian StyleReinikainen, Matti, Aki Braunschweiler, Sampsa Korpilo, Pekka Simell, and Ville Alopaeus. 2022. "Two-Step Conversion of CO2 to Light Olefins: Laboratory-Scale Demonstration and Scale-Up Considerations" ChemEngineering 6, no. 6: 96. https://doi.org/10.3390/chemengineering6060096
APA StyleReinikainen, M., Braunschweiler, A., Korpilo, S., Simell, P., & Alopaeus, V. (2022). Two-Step Conversion of CO2 to Light Olefins: Laboratory-Scale Demonstration and Scale-Up Considerations. ChemEngineering, 6(6), 96. https://doi.org/10.3390/chemengineering6060096