
Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.3. Characterization
3. Results and Discussion
3.1. Element Chemical Analysis
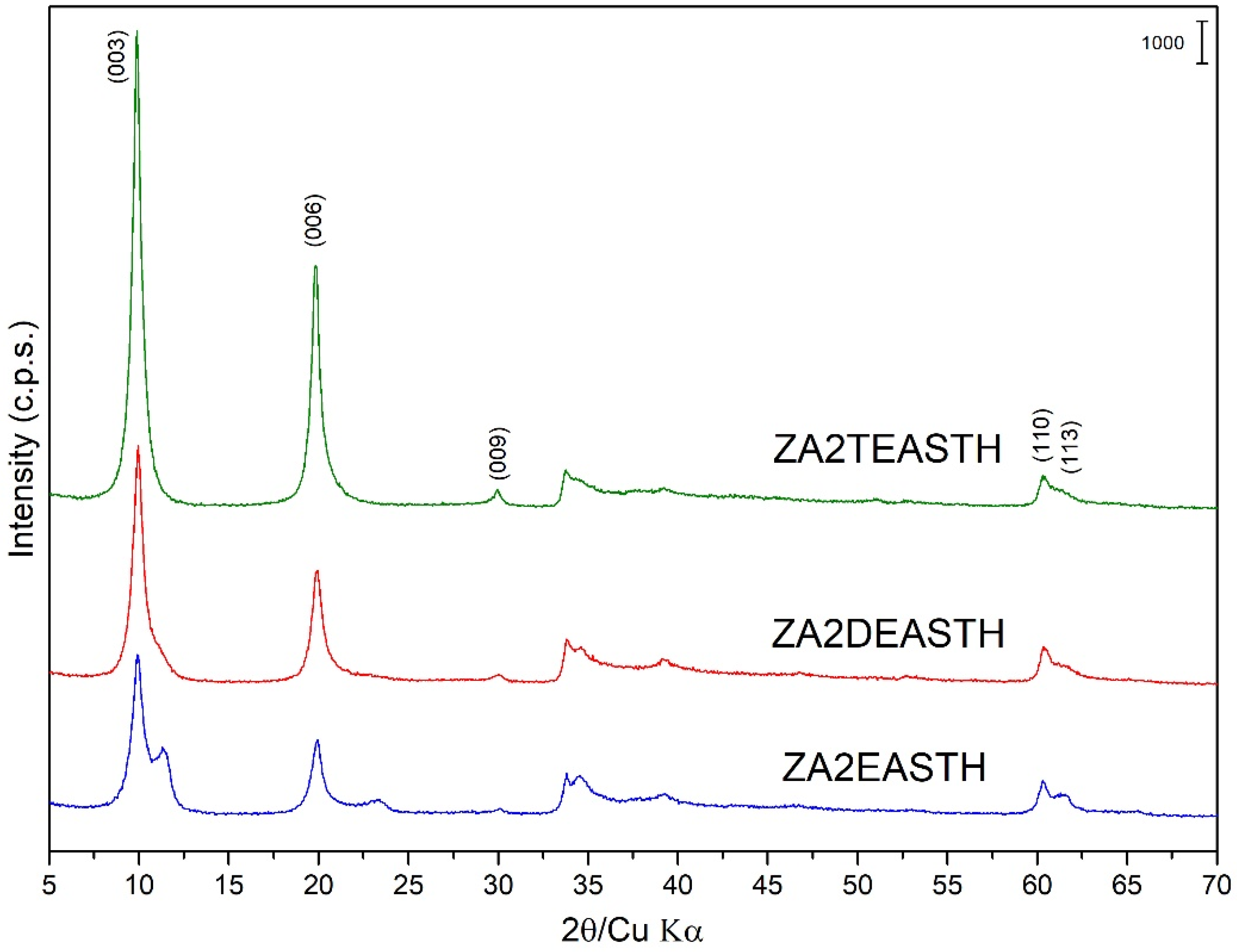
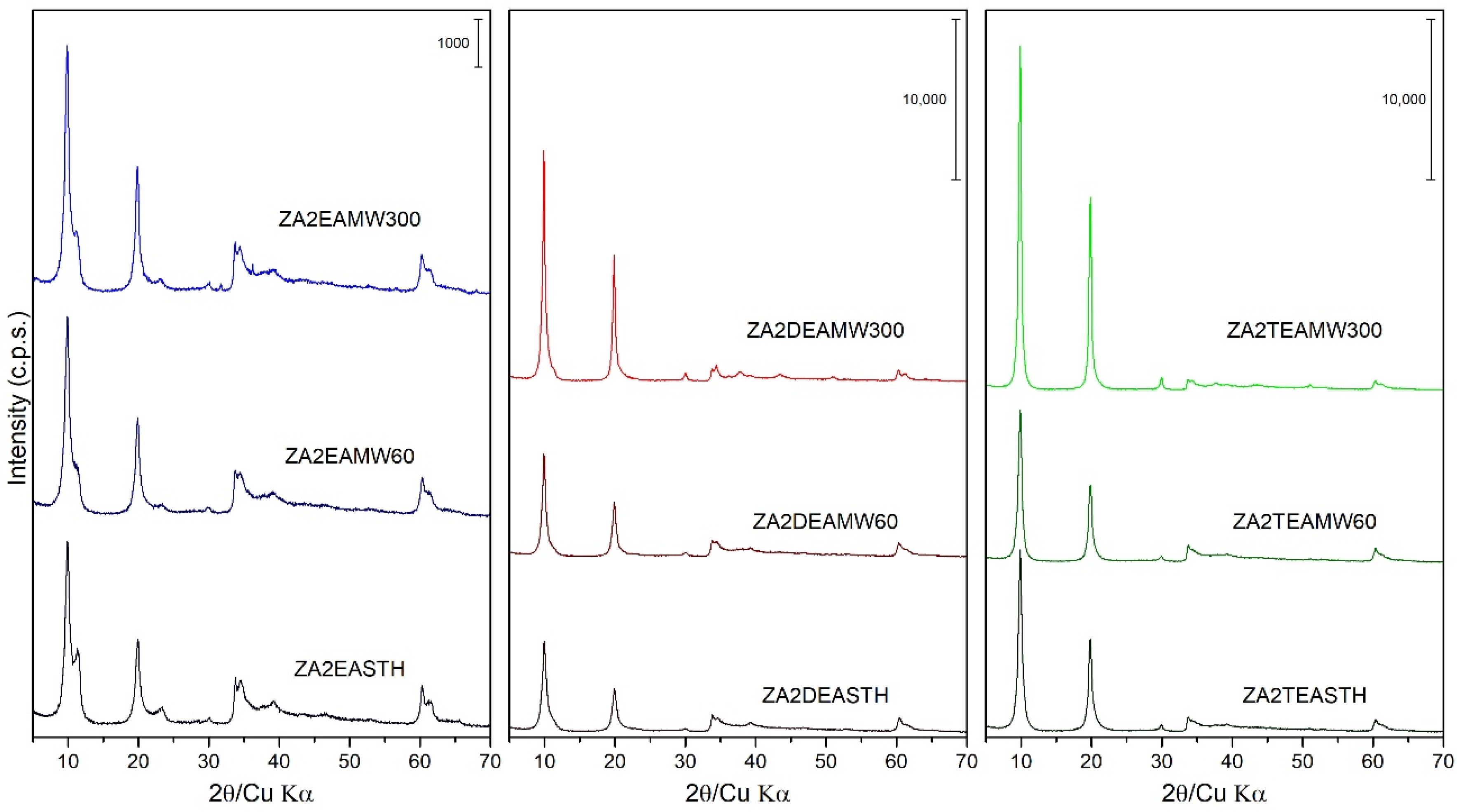
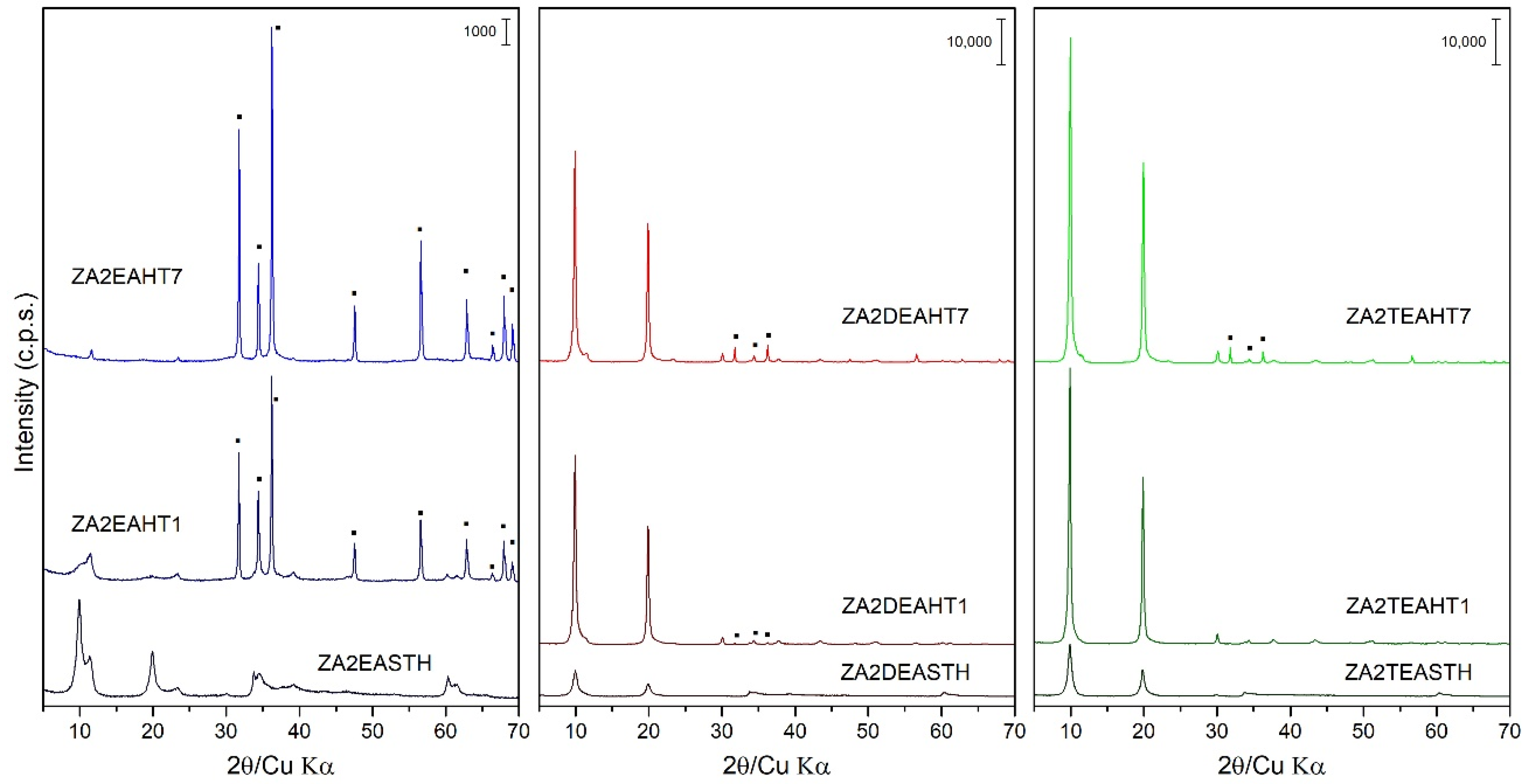
3.2. Powder X-ray Diffraction (PXRD)
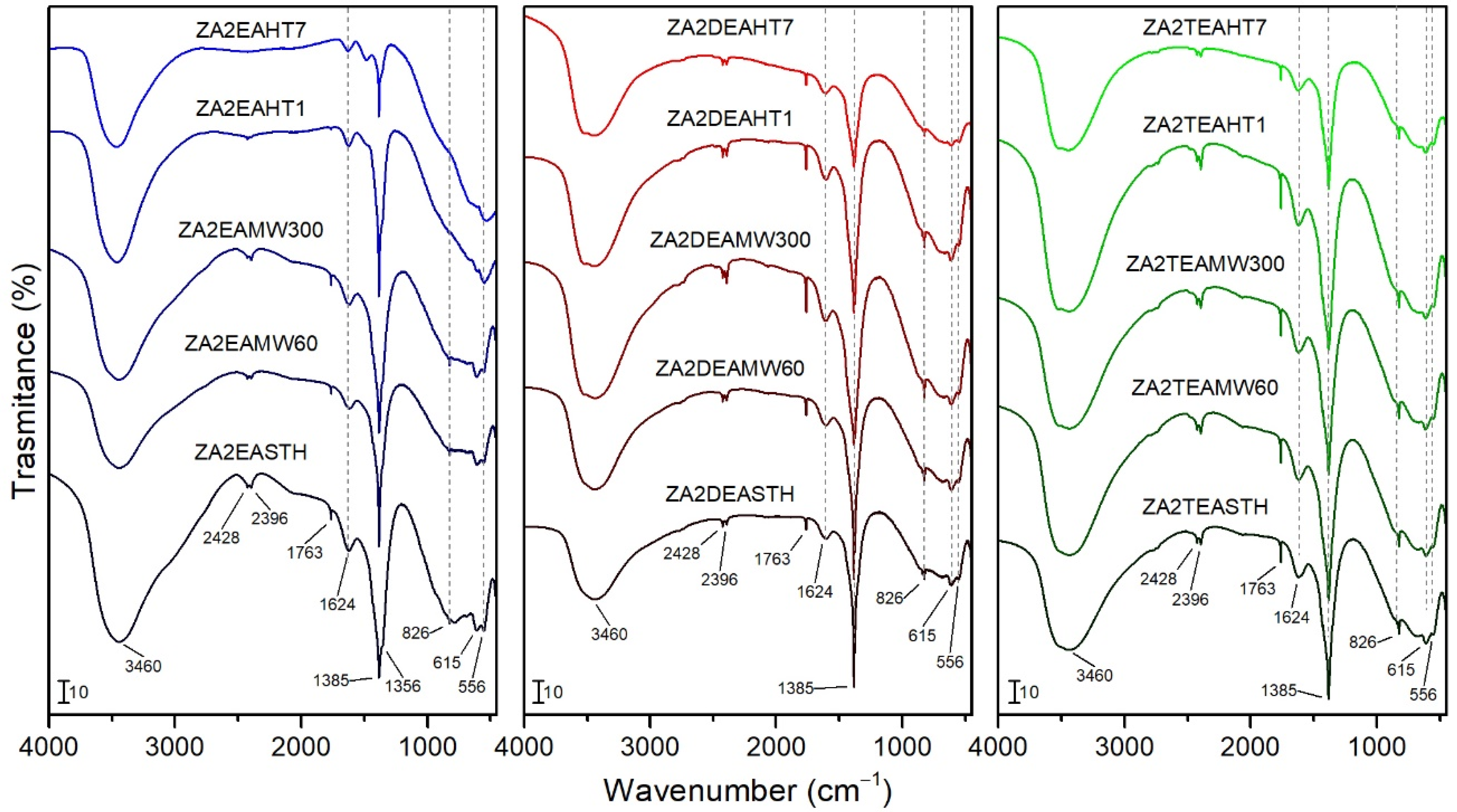
3.3. FT-IR Spectroscopy
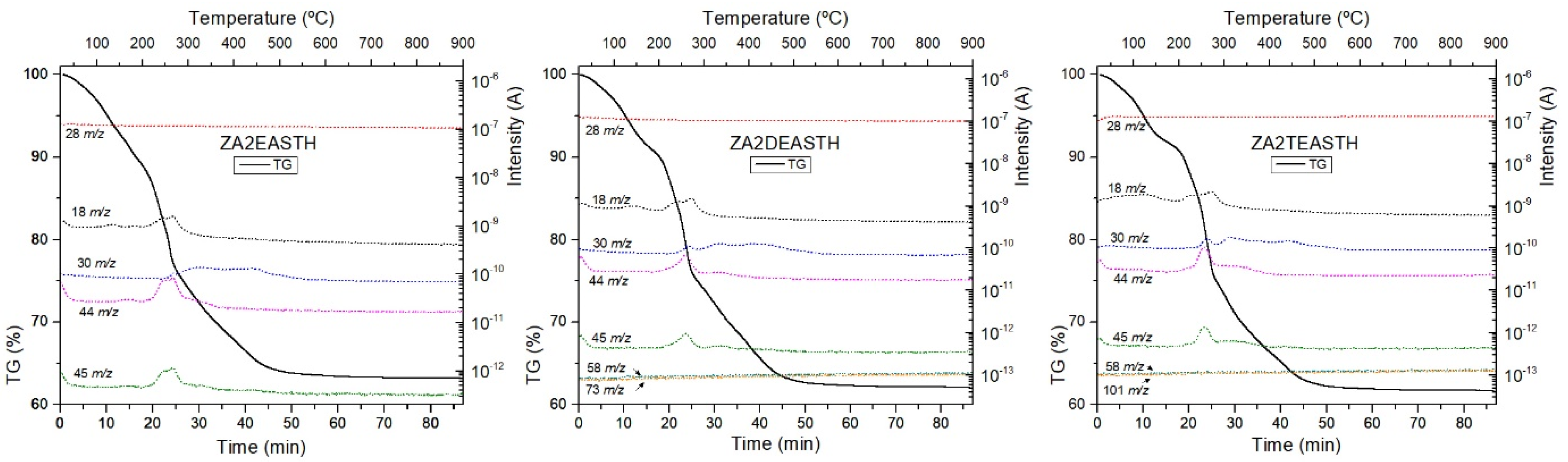
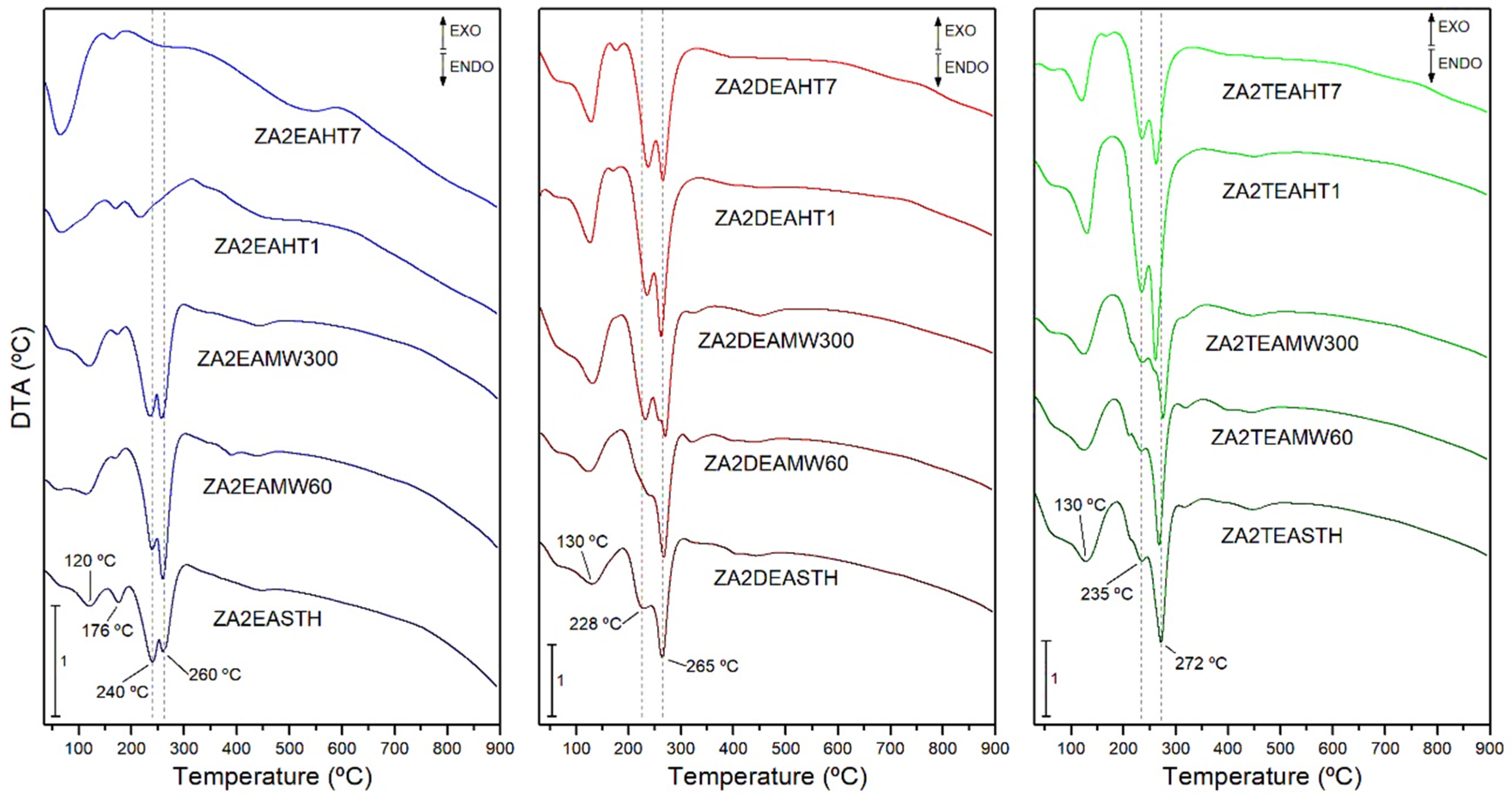
3.4. Thermal Analysis
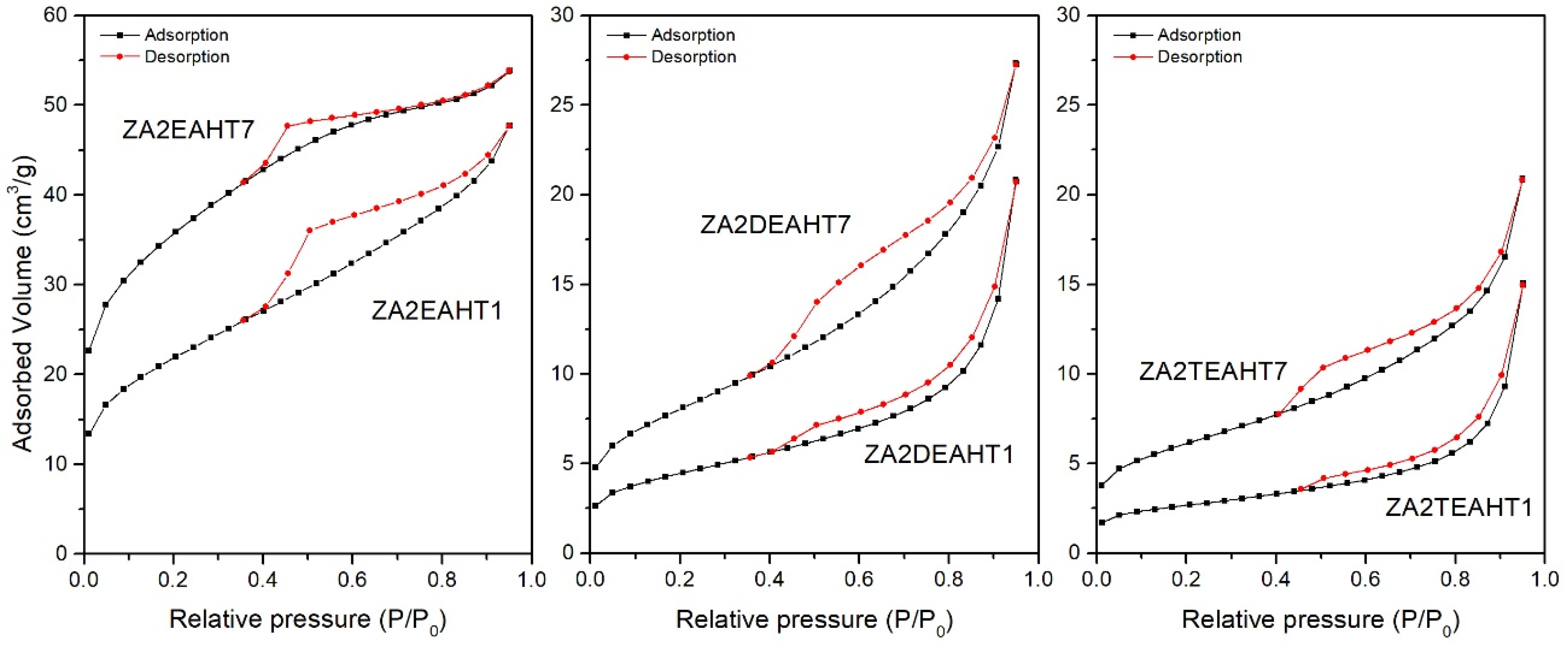
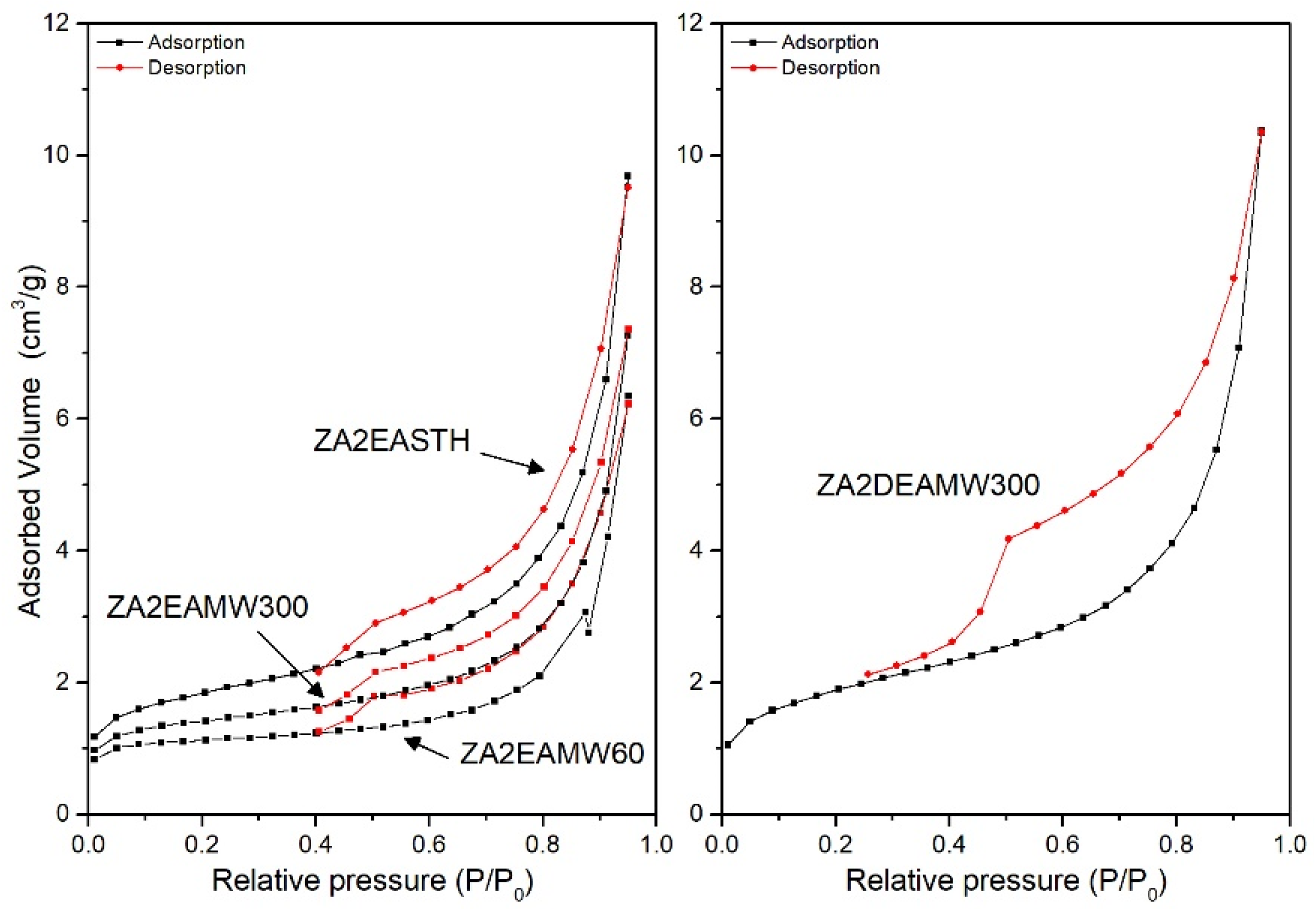
3.5. Specific Surface Area and Porosity
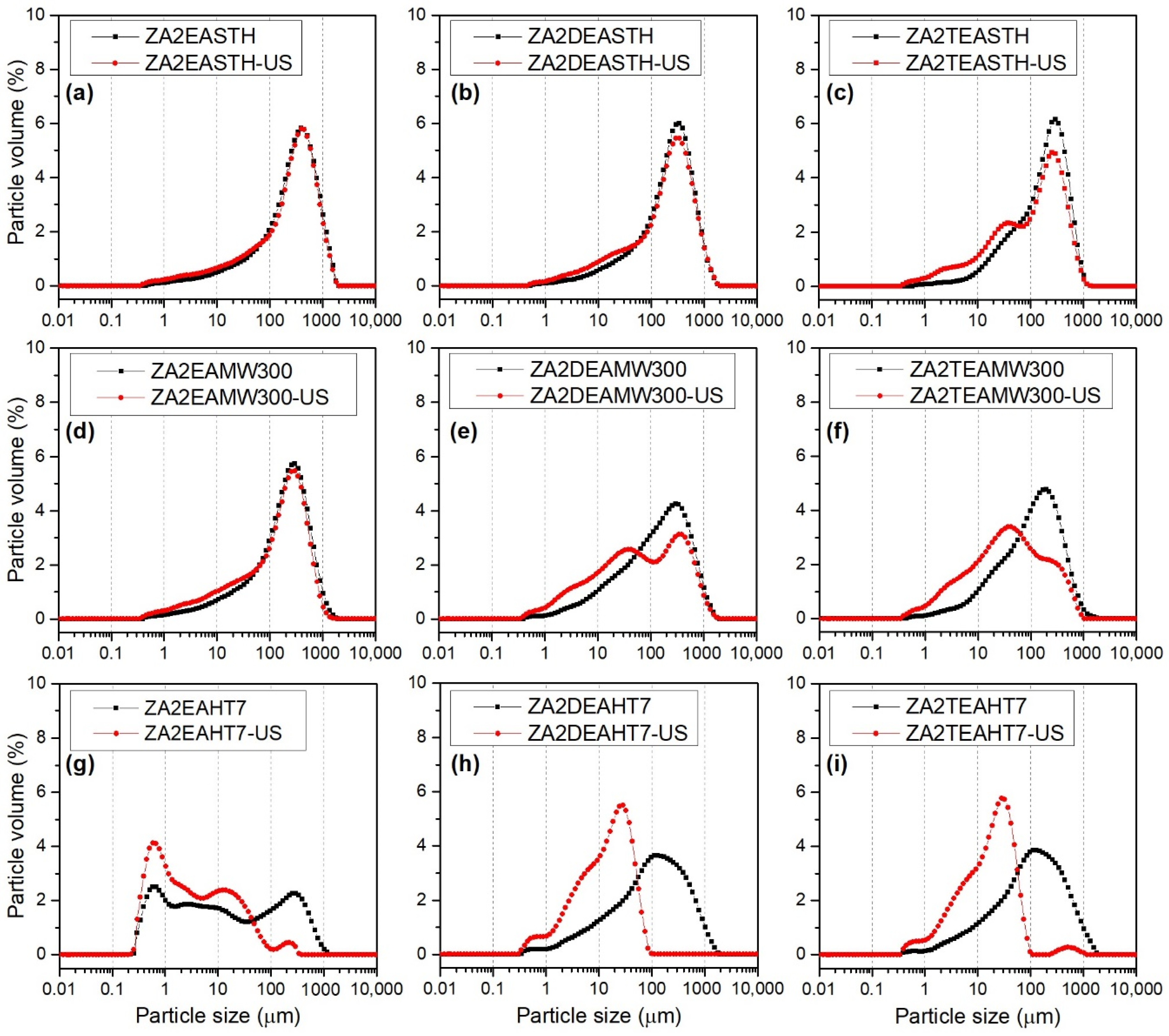
3.6. Particle Size Distribution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Rives, V.; Labajos, F.M.; Herrero, M. Layered Double Hydroxides as Nanofillers of Composites and Nanocomposite Materials Based on Polyethylene. In Polyethylene-Based Blends, Composites and Nanocomposites; Visakh, P.M., Morlanes, M.J.M., Eds.; Wiley: Beverly, MA, USA, 2015; pp. 163–200. [Google Scholar]
- Rives, V. Layered Double Hydroxides: Present and Future; NOVA Science Publishers, Inc.: New York, NY, USA, 2001; ISBN 978-1-61209-289-8. [Google Scholar]
- Wang, Q.; O’hare, D. Recent advances in the synthesis and application of layered double hydroxide (LDH) nanosheets. Chem. Rev. 2012, 112, 4124–4155. [Google Scholar] [CrossRef] [PubMed]
- Alonso-de-Linaje, V.; Mangayayam, M.C.; Tobler, D.J.; Dietmann, K.M.; Espinosa, R.; Rives, V.; Dalby, K.N. Sorption of chlorinated hydrocarbons from synthetic and natural groundwater by organo-hydrotalcites: Towards their applications as remediation nanoparticles. Chemosphere 2019, 236, 124369. [Google Scholar] [CrossRef]
- Dietmann, K.M.; Linke, T.; Trujillano, R.; Rives, V. Effect of chain length and functional group of organic anions on the retention ability of mgal-layered double hydroxides for chlorinated organic solvents. ChemEngineering 2019, 3, 89. [Google Scholar] [CrossRef]
- Alonso-de-Linaje, V.; Mangayayam, M.C.; Tobler, D.J.; Rives, V.; Espinosa, R.; Dalby, K.N. Enhanced sorption of perfluorooctane sulfonate and perfluorooctanoate by hydrotalcites. Environ. Technol. Innov. 2021, 21, 101231. [Google Scholar] [CrossRef]
- Trujillano, R.; Nájera, C.; Rives, V. Activity in the Photodegradation of 4-Nitrophenol of a Zn,Al Hydrotalcite-Like Solid and the Derived Alumina-Supported ZnO. Catalysts 2020, 10, 702. [Google Scholar] [CrossRef]
- Karásková, K.; Pacultová, K.; Jirátová, K.; Fridrichová, D.; Koštejn, M.; Obalová, L. K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx. Catalysts 2020, 10, 1134. [Google Scholar] [CrossRef]
- Li, P.; Yu, F.; Altaf, N.; Zhu, M.; Li, J.; Dai, B.; Wang, Q. Two-Dimensional Layered Double Hydroxides for Reactions of Methanation and Methane Reforming in C1 Chemistry. Materials 2018, 11, 221. [Google Scholar] [CrossRef]
- Rives, V.; Del Arco, M.; Martín, C. Layered double hydroxides as drug carriers and for controlled release of non-steroidal antiinflammatory drugs (NSAIDs): A review. J. Control. Release 2013, 169, 28–39. [Google Scholar] [CrossRef]
- Choi, G.; Choy, J. Recent progress in layered double hydroxides as a cancer theranostic nanoplatform. WIREs Nanomed. Nanobiotechnol. 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Patel, R.; Park, J.T.; Patel, M.; Dash, J.K.; Gowd, E.B.; Karpoormath, R.; Mishra, A.; Kwak, J.; Kim, J.H. Transition-metal-based layered double hydroxides tailored for energy conversion and storage. J. Mater. Chem. A 2018, 6, 12–29. [Google Scholar] [CrossRef]
- Saifullah, B.; Hussein, M.Z. Inorganic nanolayers: Structure, preparation, and biomedical applications. Int. J. Nanomed. 2015, 10, 5609–5633. [Google Scholar] [CrossRef]
- Mishra, G.; Dash, B.; Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Miyata, S. Anion-Exchange Properties of Hydrotalcite-Like Compounds. Clays Clay Miner. 1983, 31, 305–311. [Google Scholar] [CrossRef]
- Inayat, A.; Klumpp, M.; Schwieger, W. The urea method for the direct synthesis of ZnAl layered double hydroxides with nitrate as the interlayer anion. Appl. Clay Sci. 2011, 51, 452–459. [Google Scholar] [CrossRef]
- Abderrazek, K.; Frini Srasra, N.; Srasra, E. Synthesis and Characterization of [Zn-Al] Layered Double Hydroxides: Effect of the Operating Parameters. J. Chin. Chem. Soc. 2017, 64, 346–353. [Google Scholar] [CrossRef]
- Bukhtiyarova, M.V. A review on effect of synthesis conditions on the formation of layered double hydroxides. J. Solid State Chem. 2019, 269, 494–506. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Hickey, L.; Frost, R.L. The effects of synthesis pH and hydrothermal treatment on the formation of zinc aluminum hydrotalcites. J. Solid State Chem. 2004, 177, 4047–4057. [Google Scholar] [CrossRef]
- Galvão, T.L.P.; Neves, C.S.; Caetano, A.P.F.; Maia, F.; Mata, D.; Malheiro, E.; Ferreira, M.J.; Bastos, A.C.; Salak, A.N.; Gomes, J.R.B.; et al. Control of crystallite and particle size in the synthesis of layered double hydroxides: Macromolecular insights and a complementary modeling tool. J. Colloid Interface Sci. 2016, 468, 86–94. [Google Scholar] [CrossRef]
- Benito, P.; Herrero, M.; Barriga, C.; Labajos, F.M.; Rives, V. Microwave-assisted homogeneous precipitation of hydrotalcites by urea hydrolysis. Inorg. Chem. 2008, 47, 5453–5463. [Google Scholar] [CrossRef]
- He, J.; Wei, M.; Li, B.; Kang, Y.; Evans, D.G.; Duan, X. Preparation of Layered Double Hydroxides. In Layered Double Hydroxides. Structure and Bonding; Duan, X., Evans, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 119, pp. 89–119. [Google Scholar]
- Labajos, F.M.; Rives, V.; Ulibarri, M.A. Effect of hydrothermal and thermal treatments on the physicochemical properties of Mg-Al hydrotalcite-like materials. J. Mater. Sci. 1992, 27, 1546–1552. [Google Scholar] [CrossRef]
- Ezeh, C.I.; Tomatis, M.; Yang, X.; He, J.; Sun, C. Ultrasonic and hydrothermal mediated synthesis routes for functionalized Mg-Al LDH: Comparison study on surface morphology, basic site strength, cyclic sorption efficiency and effectiveness. Ultrason. Sonochem. 2018, 40, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zadaviciute, S.; Baltakys, K.; Bankauskaite, A. The effect of microwave and hydrothermal treatments on the properties of hydrotalcite. J. Therm. Anal. Calorim. 2017, 127, 189–196. [Google Scholar] [CrossRef]
- Misol, A.; Labajos, F.M.; Morato, A.; Rives, V. Synthesis of Zn,Al layered double hydroxides in the presence of amines. Appl. Clay Sci. 2020, 189, 105539. [Google Scholar] [CrossRef]
- Kooli, F.; Depège, C.; Ennaqadi, A.; De Roy, A.; Besse, J.P. Rehydration of Zn-Al layered double hydroxides. Clays Clay Miner. 1997, 45, 92–98. [Google Scholar] [CrossRef]
- De La Rosa-Guzmán, M.Á.; Guzmán-Vargas, A.; Cayetano-Castro, N.; Del Río, J.M.; Corea, M.; Martínez-Ortiz, M.D.J. Thermal stability evaluation of polystyrene-Mg/zn/Al LDH nanocomposites. Nanomaterials 2019, 9, 1528. [Google Scholar] [CrossRef]
- Drits, V.A.; Bookin, A.S. Crystal Structure and X-Ray Identification of Layered Double Hydroxides. In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; NOVA Science Publishers, Inc.: New York, NY, USA, 2001; pp. 41–100. [Google Scholar]
- Thomas, G.S.; Radha, A.V.; Kamath, P.V.; Kannan, S. Thermally induced polytype transformations among the Layered Double Hydrodides (LDHs) of Mg Zn with Al. J. Phys. Chem. B 2006, 110, 12365–12371. [Google Scholar] [CrossRef]
- Marappa, S.; Radha, S.; Kamath, P.V. Nitrate-Intercalated Layered Double Hydroxides–Structure Model, Order, and Disorder. Eur. J. Inorg. Chem. 2013, 2013, 2122–2128. [Google Scholar] [CrossRef]
- Karthikeyan, J.; Fjellvåg, H.; Bundli, S.; Sjåstad, A.O. Efficient Exfoliation of Layered Double Hydroxides; Effect of Cationic Ratio, Hydration State, Anions and Their Orientations. Materials 2021, 14, 346. [Google Scholar] [CrossRef]
- Wang, S.L.; Wang, P.C. In situ XRD and ATR-FTIR study on the molecular orientation of interlayer nitrate in Mg/Al-layered double hydroxides in water. Coll. Surf. A Physicochem. Eng. Asp. 2007, 292, 131–138. [Google Scholar] [CrossRef]
- Misol, A.; Jiménez, A.; Morato, A.; Labajos, F.M.; Rives, V. Quantification by Powder X-ray Diffraction of Metal Oxides Segregation During Formation of Layered Double Hydroxides. Eur. J. Eng. Technol. Res. 2020, 5, 1243–1248. [Google Scholar] [CrossRef]
- Brown, J.G. X-rays and Their Applications; Plenum/Ros.; Plenum Publishing Corporation: New York, NY, USA, 1966; ISBN 0-306-20021-X. [Google Scholar]
- Jenkins, R.; de Vries, J.L. Worked Examples in X-ray Analysis, 2nd ed.; Springer: New York, NY, USA, 1970; ISBN 978-1-4899-2649-4. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part A: Theory and Applications in Inorganic Chemistry, 6th ed.; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2009; ISBN 9780471743392. [Google Scholar]
- Zhang, Y.; Wang, L.; Zou, L.; Xue, D. Crystallization behaviors of hexagonal nanoplatelet MgAlCO3 layered double hydroxide. J. Cryst. Growth 2010, 312, 3367–3372. [Google Scholar] [CrossRef]
- Feng, W.; Chen, J.; Hou, C.-Y. Growth and characterization of ZnO needles. Appl. Nanosci. 2014, 4, 15–18. [Google Scholar] [CrossRef][Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Brunauer, S.; Deming, L.S.; Deming, W.E.; Teller, E. On a Theory of the van der Waals Adsorption of Gases. J. Am. Chem. Soc. 1940, 62, 1723–1732. [Google Scholar] [CrossRef]
- A Basic Guide to Particle Characterization; Malvern Instruments Limited: Malvern, UK, 2015; pp. 1–24. Available online: https://www.cif.iastate.edu/sites/default/files/uploads/Other_Inst/Particle%20Size/Particle%20Characterization%20Guide.pdf (accessed on 30 June 2022).
- A Guidebook to Particle Size Analysis. Horiba Sci. 2019, 1–32. Available online: https://www.horiba.com/aut/scientific/products/particle-characterization/particle-size-essentials-guidebook/ (accessed on 30 June 2022).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Al a | Zn a | Zn/Al b | x c | Formulae |
|---|---|---|---|---|---|
| ZA2EASTH | 8.32 | 40.27 | 2.00 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.44 H2O |
| ZA2EAMW60 | 7.98 | 38.63 | 2.00 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.48 H2O |
| ZA2EAMW300 | 7.96 | 38.30 | 1.99 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.50 H2O |
| ZA2EAHT1 | 9.84 | 46.51 | 1.95 | 0.34 | LDH * + ZnO + Al2O3 |
| ZA2EAHT7 | 11.05 | 52.01 | 1.94 | 0.34 | LDH * + ZnO + Al2O3 |
| ZA2DEASTH | 7.51 | 38.44 | 2.11 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.57 H2O |
| ZA2DEAMW60 | 7.67 | 38.87 | 2.09 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.55 H2O |
| ZA2DEAMW300 | 7.64 | 38.31 | 2.07 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.55 H2O |
| ZA2DEAHT1 | 7.73 | 38.47 | 2.05 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.51 H2O |
| ZA2DEAHT7 | 8.37 | 41.88 | 2.07 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.42 H2O |
| ZA2TEASTH | 7.64 | 37.56 | 2.03 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.55 H2O |
| ZA2TEAMW60 | 7.62 | 37.62 | 2.04 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.51 H2O |
| ZA2TEAMW300 | 7.60 | 37.39 | 2.03 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.52 H2O |
| ZA2TEAHT1 | 7.67 | 37.82 | 2.04 | 0.33 | [Zn0.67Al0.33(OH)2](NO3)0.33 · 0.50 H2O |
| ZA2TEAHT7 | 8.13 | 41.16 | 2.09 | 0.32 | [Zn0.68Al0.32(OH)2](NO3)0.32 · 0.43 H2O |
| Sample | (101) Peak Area (ZnO) a | (003) Peak Area (LDH) a | Area Ratio (101)/(003) | ZnO Content b |
|---|---|---|---|---|
| ZA2EAHT1 | 1807.0 | 1424.0 | 1.268961 | 67 |
| ZA2EAHT7 | 2776.0 | - | - | ≈100 |
| ZA2DEAHT1 | 122.1 | 18,339.8 | 0.006659 | 0.32 |
| ZA2DEAHT7 | 790.7 | 19,232.1 | 0.041114 | 2.14 |
| ZA2TEAHT1 | - | - | - | - |
| ZA2TEAHT7 | 502.6 | 26,627.0 | 0.018876 | 0.97 |
| Sample | c (Å) | a (Å) | D (Å) | Number of Stacked Layers |
|---|---|---|---|---|
| ZA2EASTH | 26.80 | 3.0697 | 111 | 12 |
| ZA2EAMW60 | 26.80 | 3.0697 | 118 | 13 |
| ZA2EAMW300 | 26.80 | 3.0720 | 135 | 15 |
| ZA2EAHT1 | - | - | - | - |
| ZA2EAHT7 | - | - | - | - |
| ZA2DEASTH | 26.67 | 3.0673 | 126 | 14 |
| ZA2DEAMW60 | 26.80 | 3.0697 | 148 | 17 |
| ZA2DEAMW300 | 26.80 | 3.0720 | 210 | 23 |
| ZA2DEAHT1 | 26.67 | 3.0743 | 284 | 32 |
| ZA2DEAHT7 | 26.80 | 3.0766 | 334 | 37 |
| ZA2TEASTH | 26.80 | 3.0697 | 150 | 17 |
| ZA2TEAMW60 | 26.80 | 3.0697 | 142 | 16 |
| ZA2TEAMW300 | 26.80 | 3.0697 | 218 | 24 |
| ZA2TEAHT1 | 26.80 | 3.0766 | 319 | 36 |
| ZA2TEAHT7 | 26.67 | 3.0743 | 322 | 36 |
| Sample | Weight Loss (%) | H2O Molecules Per Chemical Formula (n) |
|---|---|---|
| ZA2EASTH | 36.6 | 0.44 |
| ZA2EAMW60 | 36.7 | 0.48 |
| ZA2EAMW300 | 38.0 | 0.50 |
| ZA2EAHT1 | 22.6 | - |
| ZA2EAHT7 | 14.6 | - |
| ZA2DEASTH | 38.1 | 0.57 |
| ZA2DEAMW60 | 37.8 | 0.55 |
| ZA2DEAMW300 | 38.2 | 0.55 |
| ZA2DEAHT1 | 37.3 | 0.51 |
| ZA2DEAHT7 | 31.0 | 0.42 |
| ZA2TEASTH | 38.7 | 0.55 |
| ZA2TEAMW60 | 38.3 | 0.51 |
| ZA2TEAMW300 | 38.5 | 0.52 |
| ZA2TEAHT1 | 37.9 | 0.50 |
| ZA2TEAHT7 | 32.8 | 0.43 |
| Sample | SBET (m2/g) | Vpore (mm3/g) | BJH Desorption Average Pore Diameter (nm) |
|---|---|---|---|
| ZA2EASTH | 6.1 | 14.9 | 9.1 |
| ZA2EAMW60 | 3.4 | 9.8 | 9.6 |
| ZA2EAMW300 | 4.5 | 11.2 | 9.6 |
| ZA2EAHT1 | 74.3 | 73.7 | 4.6 |
| ZA2EAHT7 | 118.5 | 83.2 | 3.0 |
| ZA2DEASTH | - | - | - |
| ZA2DEAMW60 | - | - | - |
| ZA2DEAMW300 | 6.4 | 16.0 | 6.9 |
| ZA2DEAHT1 | 15.3 | 32.2 | 7.6 |
| ZA2DEAHT7 | 28.2 | 42.2 | 5.3 |
| ZA2TEASTH | - | - | - |
| ZA2TEAMW60 | - | - | - |
| ZA2TEAMW300 | - | - | - |
| ZA2TEAHT1 | 9.0 | 23.3 | 9.9 |
| ZA2TEAHT7 | 21.0 | 32.4 | 5.7 |
| Sample | D[4,3] Before Sonication | D[4,3] After Sonication |
|---|---|---|
| ZA2EASTH | 428 | 399 |
| ZA2EAMW60 | 280 | 258 |
| ZA2EAMW300 | 298 | 248 |
| ZA2EAHT1 | 327 | 230 |
| ZA2EAHT7 | 114 | 16 |
| ZA2DEASTH | 347 | 324 |
| ZA2DEAMW60 | 268 | 263 |
| ZA2DEAMW300 | 275 | 210 |
| ZA2DEAHT1 | 272 | 94 |
| ZA2DEAHT7 | 232 | 22 |
| ZA2TEASTH | 274 | 212 |
| ZA2TEAMW60 | 221 | 219 |
| ZA2TEAMW300 | 201 | 112 |
| ZA2TEAHT1 | 253 | 79 |
| ZA2TEAHT7 | 231 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misol, A.; Jiménez, A.; Labajos, F.M. Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides. ChemEngineering 2022, 6, 53. https://doi.org/10.3390/chemengineering6040053
Misol A, Jiménez A, Labajos FM. Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides. ChemEngineering. 2022; 6(4):53. https://doi.org/10.3390/chemengineering6040053
Chicago/Turabian StyleMisol, Alexander, Alejandro Jiménez, and Francisco M. Labajos. 2022. "Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides" ChemEngineering 6, no. 4: 53. https://doi.org/10.3390/chemengineering6040053
APA StyleMisol, A., Jiménez, A., & Labajos, F. M. (2022). Use of Ethylamine, Diethylamine and Triethylamine in the Synthesis of Zn,Al Layered Double Hydroxides. ChemEngineering, 6(4), 53. https://doi.org/10.3390/chemengineering6040053